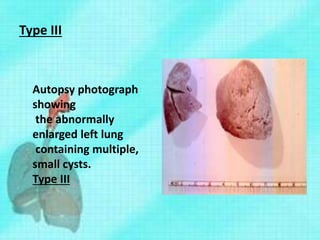
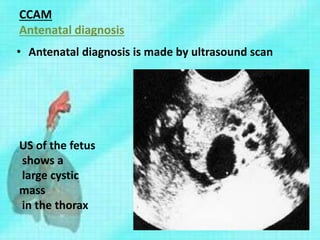
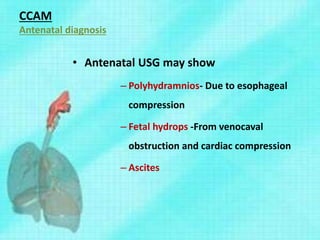
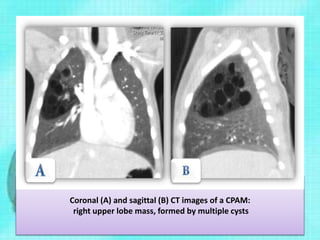
Downloaded 70 times








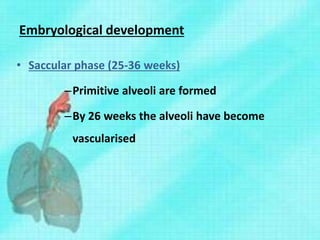












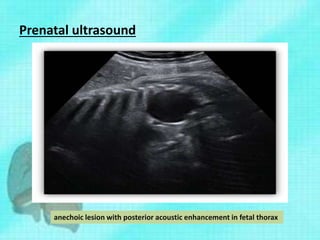
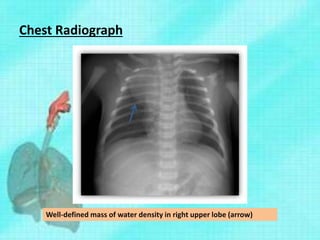
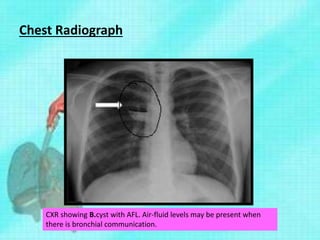
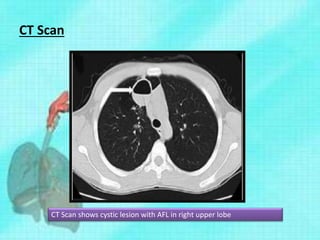
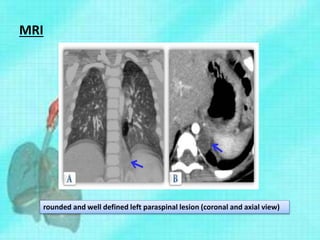
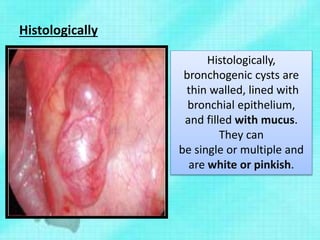






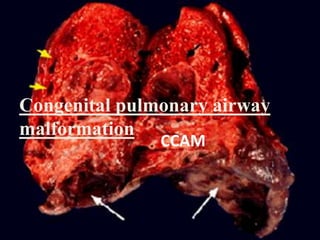

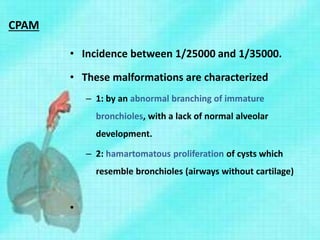


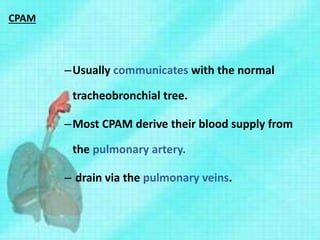






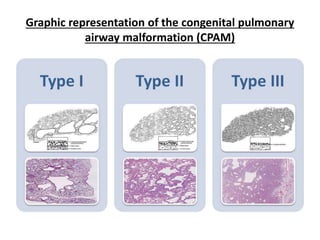

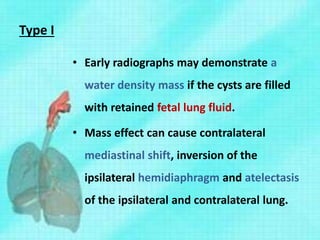
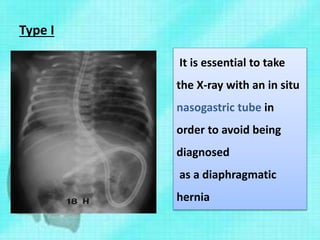
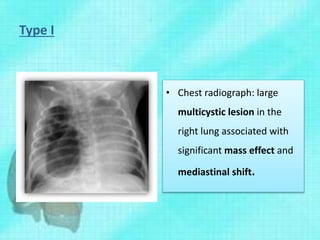

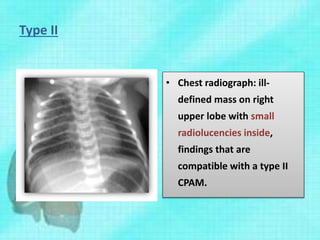


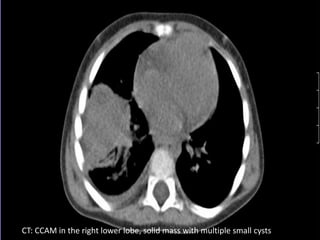

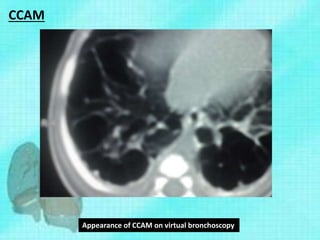
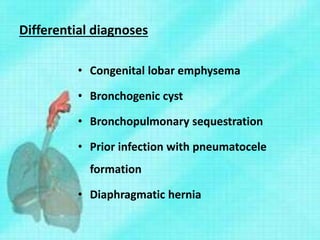
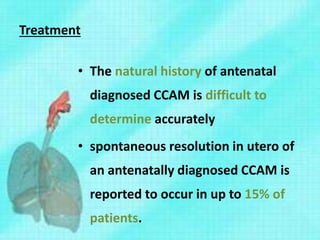









This document discusses congenital lung malformations, including bronchogenic cysts and congenital pulmonary airway malformations (CPAM). It defines these conditions, describes their embryological development, classification, clinical presentation, diagnosis and treatment. Bronchogenic cysts are abnormal budding of the tracheal diverticulum that can cause compression symptoms. CPAM is characterized by abnormal bronchiole branching and cyst formation. It discusses Stocker's classification of CPAM types based on cyst size and associated risks. Prenatal ultrasound and CT are used to diagnose these conditions. Surgical resection is the primary treatment.













































![Development_of_lung_and_disorders_part_1[2].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/developmentoflunganddisorderspart12-251218043139-55070ef1-thumbnail.jpg?width=640&height=640&fit=bounds)
























![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)








![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)







