Download free for 30 days
Sign in
Upload
Language (EN)
Support
Business
Mobile
Social Media
Marketing
Technology
Art & Photos
Career
Design
Education
Presentations & Public Speaking
Government & Nonprofit
Healthcare
Internet
Law
Leadership & Management
Automotive
Engineering
Software
Recruiting & HR
Retail
Sales
Services
Science
Small Business & Entrepreneurship
Food
Environment
Economy & Finance
Data & Analytics
Investor Relations
Sports
Spiritual
News & Politics
Travel
Self Improvement
Real Estate
Entertainment & Humor
Health & Medicine
Devices & Hardware
Lifestyle
Change Language
Language
English
Español
Português
Français
Deutsche
Cancel
Save
Submit search
EN
Uploaded by
Beckett Hsieh
PPTX, PDF
5,141 views
張偉豪-擺脫墨菲定律魔咒的迷思-Logistc迴歸-20160602
AI-enhanced description
本文件讨论二元罗吉斯回归分析及其应用,涵盖了基本概念、步骤及结果解释。文中比较了传统回归与罗吉斯回归之间的差异,并提供了如何构建模型及预测心脏病发生概率的示例。最后,分析了模型的适配性和分类的准确性。
Education
◦
Read more
3
Save
Share
Embed
Embed presentation
Download
Downloaded 116 times
1
/ 58
2
/ 58
3
/ 58
4
/ 58
5
/ 58
6
/ 58
7
/ 58
8
/ 58
9
/ 58
10
/ 58
11
/ 58
12
/ 58
13
/ 58
14
/ 58
15
/ 58
16
/ 58
17
/ 58
18
/ 58
19
/ 58
20
/ 58
21
/ 58
22
/ 58
23
/ 58
24
/ 58
25
/ 58
26
/ 58
27
/ 58
28
/ 58
29
/ 58
30
/ 58
31
/ 58
32
/ 58
33
/ 58
34
/ 58
35
/ 58
36
/ 58
37
/ 58
38
/ 58
39
/ 58
40
/ 58
41
/ 58
42
/ 58
43
/ 58
44
/ 58
45
/ 58
46
/ 58
47
/ 58
48
/ 58
49
/ 58
50
/ 58
51
/ 58
52
/ 58
53
/ 58
54
/ 58
55
/ 58
56
/ 58
57
/ 58
58
/ 58
More Related Content
PPTX
SPSS從0開始-三星統計張偉豪
by
Beckett Hsieh
PPTX
Sclerostin edit 2
by
Irsalanasif
PPTX
三星統計張偉豪-統計4超人-SPSS,SEM,HLM,PLS
by
Beckett Hsieh
PDF
看不見的消費者 Google關鍵字廣告實作-三星統計謝章升
by
Beckett Hsieh
PDF
謝章升-Google Analytics網站流量分析入門實作
by
Beckett Hsieh
PDF
FB廣告入門
by
Beckett Hsieh
PDF
SmartPLS3.0偏最小平方法教材2015版-三星統計張偉豪
by
Beckett Hsieh
PDF
大數據行銷應用與SPSS實作-三星統計謝章升
by
Beckett Hsieh
SPSS從0開始-三星統計張偉豪
by
Beckett Hsieh
Sclerostin edit 2
by
Irsalanasif
三星統計張偉豪-統計4超人-SPSS,SEM,HLM,PLS
by
Beckett Hsieh
看不見的消費者 Google關鍵字廣告實作-三星統計謝章升
by
Beckett Hsieh
謝章升-Google Analytics網站流量分析入門實作
by
Beckett Hsieh
FB廣告入門
by
Beckett Hsieh
SmartPLS3.0偏最小平方法教材2015版-三星統計張偉豪
by
Beckett Hsieh
大數據行銷應用與SPSS實作-三星統計謝章升
by
Beckett Hsieh
Viewers also liked
PDF
網路與社群行銷管理師證照演講-三星統計謝章升-20150303
by
Beckett Hsieh
PDF
中介與調節效果當代分析手法大解秘-Amos與Mplus為例-三星統計張偉豪
by
Beckett Hsieh
PDF
SEM結構方程模型-Amos多群組比較-張偉豪-20150803
by
Beckett Hsieh
PDF
統計的力量-SPSS的25種方法實戰2014版-三星統計張偉豪20141119
by
Beckett Hsieh
PDF
SEM結構方程模型與Amos基礎班講義-三星統計張偉豪
by
Beckett Hsieh
PPTX
To be master or slave of statistics
by
Beckett Hsieh
PPTX
教材摘要版 -Big data-海量資料的資料採礦方法-三星課程網陳景祥顧問-20130521
by
Beckett Hsieh
PDF
看不見的消費者-網站設計-FB與關鍵字廣告-流量分析-三星統計謝章升
by
Beckett Hsieh
PDF
十分鐘讓程式人搞懂雲端平台與技術
by
鍾誠 陳鍾誠
PPTX
SEM結構方程模型與Amos-潛在成長模型-三星統計張偉豪
by
Beckett Hsieh
PDF
R統計分析與圖形藝術ggplot2-謝雨豆-三星統計-20140221
by
Beckett Hsieh
PPT
結構方程模式於期刊論文研究的應用-三星統計張偉豪-20131007
by
Beckett Hsieh
PDF
原來量化研究的問卷與分析是這樣簡單-三星統計謝章升-20140721
by
Beckett Hsieh
PDF
公開版-看不見的消費者-Facebook廣告與google關鍵字廣告-三星統計謝章升-20141013
by
Beckett Hsieh
PDF
Hiiir 營銷講座 營運App的二三事 創造領先App 的必勝手冊 (分享版)
by
Hiiir Lab
PDF
SEM與LISREL基礎班講義20130112-三星統計張偉豪顧問
by
Beckett Hsieh
PDF
以產出為導向-SPSS,SEM,PLS-在學術論文的應用-三星統計謝章升-20131219
by
Beckett Hsieh
PDF
HLM階層線性模型基礎班-三星統計張偉豪
by
Beckett Hsieh
PPTX
10種網路新手快速簡單的賺錢方法
by
bahn hong
PDF
SEM與Amos應用班系列一中介與干擾變數-三星統計張偉豪
by
Beckett Hsieh
網路與社群行銷管理師證照演講-三星統計謝章升-20150303
by
Beckett Hsieh
中介與調節效果當代分析手法大解秘-Amos與Mplus為例-三星統計張偉豪
by
Beckett Hsieh
SEM結構方程模型-Amos多群組比較-張偉豪-20150803
by
Beckett Hsieh
統計的力量-SPSS的25種方法實戰2014版-三星統計張偉豪20141119
by
Beckett Hsieh
SEM結構方程模型與Amos基礎班講義-三星統計張偉豪
by
Beckett Hsieh
To be master or slave of statistics
by
Beckett Hsieh
教材摘要版 -Big data-海量資料的資料採礦方法-三星課程網陳景祥顧問-20130521
by
Beckett Hsieh
看不見的消費者-網站設計-FB與關鍵字廣告-流量分析-三星統計謝章升
by
Beckett Hsieh
十分鐘讓程式人搞懂雲端平台與技術
by
鍾誠 陳鍾誠
SEM結構方程模型與Amos-潛在成長模型-三星統計張偉豪
by
Beckett Hsieh
R統計分析與圖形藝術ggplot2-謝雨豆-三星統計-20140221
by
Beckett Hsieh
結構方程模式於期刊論文研究的應用-三星統計張偉豪-20131007
by
Beckett Hsieh
原來量化研究的問卷與分析是這樣簡單-三星統計謝章升-20140721
by
Beckett Hsieh
公開版-看不見的消費者-Facebook廣告與google關鍵字廣告-三星統計謝章升-20141013
by
Beckett Hsieh
Hiiir 營銷講座 營運App的二三事 創造領先App 的必勝手冊 (分享版)
by
Hiiir Lab
SEM與LISREL基礎班講義20130112-三星統計張偉豪顧問
by
Beckett Hsieh
以產出為導向-SPSS,SEM,PLS-在學術論文的應用-三星統計謝章升-20131219
by
Beckett Hsieh
HLM階層線性模型基礎班-三星統計張偉豪
by
Beckett Hsieh
10種網路新手快速簡單的賺錢方法
by
bahn hong
SEM與Amos應用班系列一中介與干擾變數-三星統計張偉豪
by
Beckett Hsieh
More from Beckett Hsieh
PPTX
謝章升-Google Analytics進階
by
Beckett Hsieh
PPTX
謝章升-Google Analytics入門
by
Beckett Hsieh
PDF
謝章升-Google我的商家課程講義
by
Beckett Hsieh
PPTX
謝章升-Google Analytics網站流量分析入門課程講義
by
Beckett Hsieh
PDF
謝章升-個人品牌如何知識變現
by
Beckett Hsieh
PPTX
謝章升-數位行銷基本力
by
Beckett Hsieh
PDF
GTM(Google Tag Manager)懶人包-謝章升
by
Beckett Hsieh
PDF
Google廣告入門實作-三星統計謝章升-20171031
by
Beckett Hsieh
PDF
參考講義展示版-丘祐瑋-20170617-機器學習python入門者課程
by
Beckett Hsieh
PDF
謝章升-演講-大數據行銷-網路廣告解讀-20170327
by
Beckett Hsieh
PDF
謝章升-演講-Ga網站流量分析入門-20170410
by
Beckett Hsieh
PDF
謝章升-演講-拉新與熟客經營-再行銷廣告與EDM-20170413
by
Beckett Hsieh
PDF
謝章升-演講-WordPress ORG建構網頁入門-20170424
by
Beckett Hsieh
PDF
演講-Meta analysis in medical research-張偉豪
by
Beckett Hsieh
PPTX
品牌管理師證照重點複習-謝章升-20151221
by
Beckett Hsieh
PPTX
整合行銷管理師總複習-20150927
by
Beckett Hsieh
PDF
演講-DEA資料包絡法在學術論文的應用-台師大方進義-三星統計-20141119
by
Beckett Hsieh
PDF
演講-AHP與ANP在學術論文的應用-許旭昇-三星課程網
by
Beckett Hsieh
謝章升-Google Analytics進階
by
Beckett Hsieh
謝章升-Google Analytics入門
by
Beckett Hsieh
謝章升-Google我的商家課程講義
by
Beckett Hsieh
謝章升-Google Analytics網站流量分析入門課程講義
by
Beckett Hsieh
謝章升-個人品牌如何知識變現
by
Beckett Hsieh
謝章升-數位行銷基本力
by
Beckett Hsieh
GTM(Google Tag Manager)懶人包-謝章升
by
Beckett Hsieh
Google廣告入門實作-三星統計謝章升-20171031
by
Beckett Hsieh
參考講義展示版-丘祐瑋-20170617-機器學習python入門者課程
by
Beckett Hsieh
謝章升-演講-大數據行銷-網路廣告解讀-20170327
by
Beckett Hsieh
謝章升-演講-Ga網站流量分析入門-20170410
by
Beckett Hsieh
謝章升-演講-拉新與熟客經營-再行銷廣告與EDM-20170413
by
Beckett Hsieh
謝章升-演講-WordPress ORG建構網頁入門-20170424
by
Beckett Hsieh
演講-Meta analysis in medical research-張偉豪
by
Beckett Hsieh
品牌管理師證照重點複習-謝章升-20151221
by
Beckett Hsieh
整合行銷管理師總複習-20150927
by
Beckett Hsieh
演講-DEA資料包絡法在學術論文的應用-台師大方進義-三星統計-20141119
by
Beckett Hsieh
演講-AHP與ANP在學術論文的應用-許旭昇-三星課程網
by
Beckett Hsieh
張偉豪-擺脫墨菲定律魔咒的迷思-Logistc迴歸-20160602
1.
擺脫墨菲定律魔咒的迷思 SPSS二元羅吉斯迴歸分析 張偉豪 三星統計服務有限公司 執行長 Amos 亞洲一哥
2.
參考書目 http://www.semsoeasy.com.tw/
3.
大綱 ▪ What is
logistic regression? ▪ Logistic regression step-by-step ▪ How to interpret the results of logistic regression
4.
什麼是羅吉斯迴歸? ▪ 羅吉斯迴歸分析一般應用在應變數 (Binary var.)與自變數之間的關係為非 線性。 –
年齡高低與有無CHD的關係 – 糖份攝取量與有無糖尿病的關係 – 房貸金額與核準信用卡與否的關係 – 家庭支持與憂鬱症治癒的關係 – 刑期長短與假釋出獄後是否回籠的關係 – 行車速度與車禍時輕傷或重傷的關係 – 醫生經驗與開刀成功與否的關係
5.
二元 羅吉斯迴歸 Binary Logistic
Regression ▪ 結果變數(Y)必須是二分類變數 ▪ 不需符合一般多變量的嚴格假設及較具強靭性 ▪ 與區別分析同樣具有正確的統計檢定能力及整合 非線性影響的能力 ▪ 能應用於各種範圍的特徵 ▪ 適合用於建構決策模型
6.
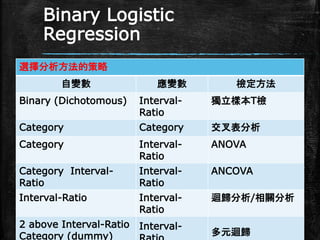
Binary Logistic Regression 選擇分析方法的策略 自變數 應變數
檢定方法 Binary (Dichotomous) Interval- Ratio 獨立樣本T檢 Category Category 交叉表分析 Category Interval- Ratio ANOVA Category Interval- Ratio Interval- Ratio ANCOVA Interval-Ratio Interval- Ratio 迴歸分析/相關分析 2 above Interval-Ratio Category (dummy) Interval- 多元迴歸
7.
二元 羅吉斯迴歸? ▪ 為何要用羅吉斯迴歸,而不直接用傳統的迴 歸分析? –
直接用傳統迴歸(最小平方法),會有什麼問題? – Y = β0 +β1 X1+ β2 X2 + ε
8.
為何不用線性迴歸? ▪ 簡單迴歸是1個連續自變數預測另1個連 續自變數 ▪ 多元迴歸只是多了幾個自變數的迴歸 ▪
執行傳統的線性迴歸會有以下幾個問題 ▪ Y為二分類變數,所以Y不會符合常態分配 ▪ 預測值可能超過1或小於0,這會違反我們機率的 定義(0~1) ▪ 機率通常不會是線性分佈 ▪ 誤差項不同質(自變數與應變數會有不同的 變異數)
9.
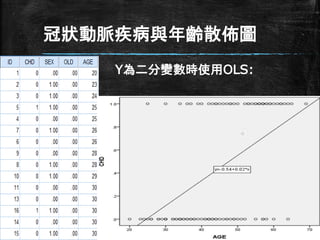
冠狀動脈疾病與年齡散佈圖 Y為二分變數時使用OLS:
10.
連續 VS. 類別變數 ▪
一般線性模型 – Y = β0 +β1 X1+ β2 X2 + ε ▪ 自變數(X) – 連續變數:年齡,身高等,數值帶入 – 類別變數:性別,教育程度等,虛擬變數帶入 ▪ 依變數(Y) – 連續變數:消費金額,時間等,數值帶入 – 類別變數:yes/no (1/0),虛擬變數帶入
11.
線性模型? ▪ Y=CHD為是否有冠狀動脈疾病 ▪ Y的值為(0)沒有冠狀動脈疾病,
(1)有冠 狀動脈疾病 ▪ 線性迴歸方程式如下 – CHD = β0 +β1 *age + ε ▪ 分析結果: CHD = -.54 +.02 *age
12.
結果解讀 ▪ 依變數CHD為二分類變數,因此值只在 0和1之間變動 ▪ 這也可以解釋成,有CHD的可能性 P(CHD=1),以P表示 ▪
結果表示為 – P(CHD=1) = P = -.54 +.02 *age ▪ 年齡每增加1歲,得CHD的可能性增加2%
13.
線性模型的問題 ▪ 機率(probability) 介於0<P<1 ▪
資料中年齡的範圍 20~69歲 ▪ 40歲的人得CHD的機率 – P = -.54 +.02 *40 = .26 ▪ 若20歲或80歲的人得CHD機率如何? – P = -.54 +.02 *20 = -.14 – P = -.54 +.02 *80= 1.06
14.
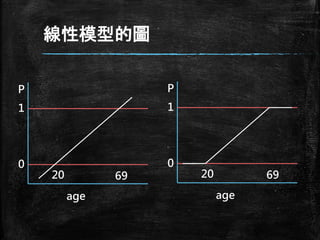
線性模型的圖 age P 0 1 20 69 age P 0 1 20 69
15.
固定P<1且P>0 ▪ 分析中必需設定P值, 並使P值在
0<P<1之 間 ▪ P值為age的函數p=f(age),但不適用線性模 型 ▪ 什麼函數f(*)會永遠滿足預測的結果 ▪ f(*)需滿足兩件事 – 它必須永遠是正的(P>0) – 它必須永遠是小於1的(P<1)
16.
兩步驟! 1. P值永遠是正的 (P>0) P=exp(β0
+β1 *age ) 2. P值永遠小於1 (P<1) P= exp(β0 +β1 age) exp(β0 +β1 age)+𝟏
17.
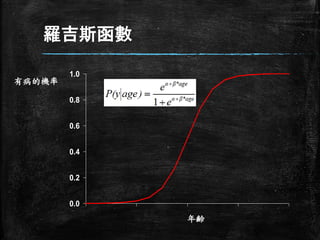
線性概念仍是存在的 ▪ 前面的表示式只是做了些數學運算,此數 學式可以被重寫成: ln( 𝑷 𝟏−𝑷 )= (β0
+β1 age) ▪ 即使年齡與CHD不是線性關係,經過簡單 的轉換也可以變成線性模型 ▪ 以上的模型就是 Logistics regression model
18.
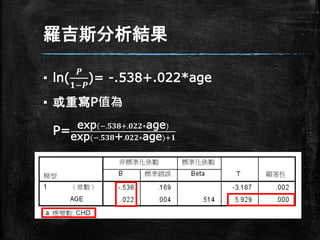
羅吉斯分析結果 ▪ ln( 𝑷 𝟏−𝑷 )= -.538+.022*age ▪
或重寫P值為 P= exp(−.𝟓𝟑𝟖+.𝟎𝟐𝟐∗age) exp(−.𝟓𝟑𝟖+.𝟎𝟐𝟐∗age)+𝟏
19.
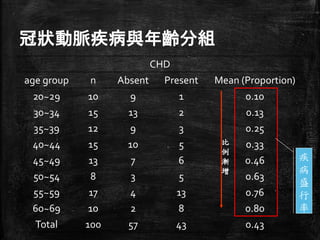
冠狀動脈疾病與年齡分組 CHD age group n
Absent Present Mean (Proportion) 20~29 10 9 1 0.10 30~34 15 13 2 0.13 35~39 12 9 3 0.25 40~44 15 10 5 0.33 45~49 13 7 6 0.46 50~54 8 3 5 0.63 55~59 17 4 13 0.76 60~69 10 2 8 0.80 Total 100 57 43 0.43 疾 病 盛 行 率 比 例 漸 增
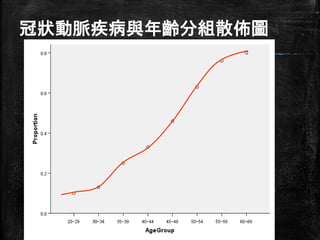
20.
冠狀動脈疾病與年齡分組散佈圖 http://www.semsoeasy.com.tw/
21.
羅吉斯函數 0.0 0.2 0.4 0.6 0.8 1.0 有病的機率 年齢
22.
Example: 冠狀動脈疾病與年齡 ▪ N=100 ▪
CHD(0) : 沒有冠狀動脈疾病 CHD(1) : 有冠狀動脈疾病 這兩個選項必須是互斥的二分變數 ▪ AGE=年齡 ▪ SEX=性別:男(1),女(0) ▪ OLD=46歲以上(1), 45歲以下(0) ▪ 左圖15/100樣本
23.
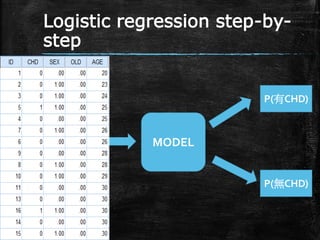
Logistic regression step-by- step MODEL P(有CHD) P(無CHD)
24.
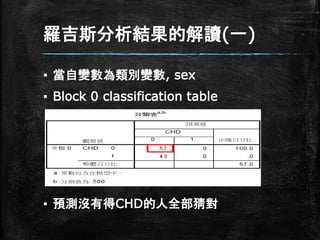
羅吉斯分析結果的解讀(一) ▪ 當自變數為類別變數, sex ▪
Block 0 classification table ▪ 預測沒有得CHD的人全部猜對
25.
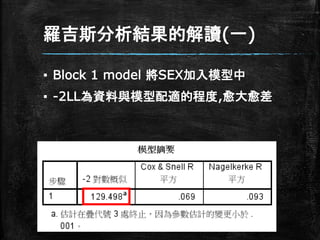
羅吉斯分析結果的解讀(一) ▪ Block 1
model 將SEX加入模型中 ▪ -2LL為資料與模型配適的程度,愈大愈差
26.
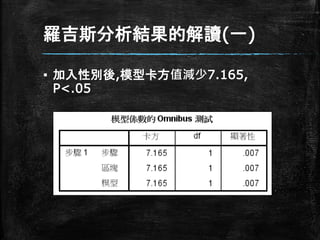
羅吉斯分析結果的解讀(一) ▪ 加入性別後,模型卡方值減少7.165, P<.05
27.
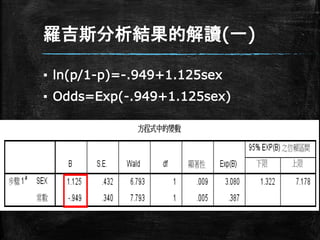
羅吉斯分析結果的解讀(一) ▪ ln(p/1-p)=-.949+1.125sex ▪ Odds=Exp(-.949+1.125sex)
28.
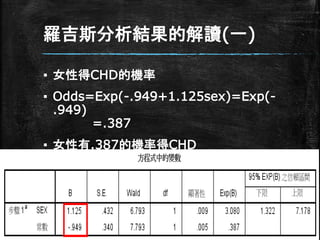
羅吉斯分析結果的解讀(一) ▪ 女性得CHD的機率 ▪ Odds=Exp(-.949+1.125sex)=Exp(- .949) =.387 ▪
女性有.387的機率得CHD
29.
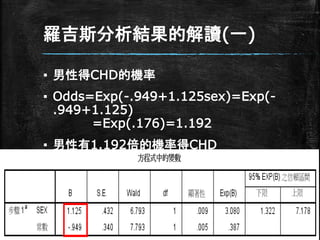
羅吉斯分析結果的解讀(一) ▪ 男性得CHD的機率 ▪ Odds=Exp(-.949+1.125sex)=Exp(- .949+1.125) =Exp(.176)=1.192 ▪
男性有1.192倍的機率得CHD
30.
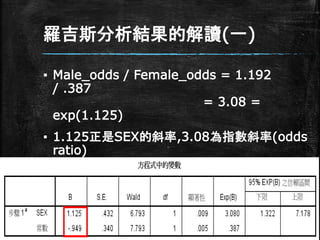
羅吉斯分析結果的解讀(一) ▪ Male_odds /
Female_odds = 1.192 / .387 = 3.08 = exp(1.125) ▪ 1.125正是SEX的斜率,3.08為指數斜率(odds ratio) ▪ 男性比女性多了3.08倍的機會得CHD
31.
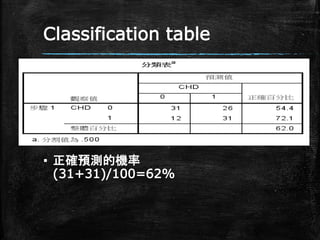
Classification table ▪ 正確預測的機率 (31+31)/100=62%
32.
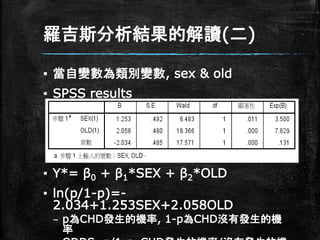
羅吉斯分析結果的解讀(二) ▪ 當自變數為類別變數, sex
& old ▪ SPSS results ▪ Y*= β0 + β1*SEX + β2*OLD ▪ ln(p/1-p)=- 2.034+1.253SEX+2.058OLD – p為CHD發生的機率, 1-p為CHD沒有發生的機 率
33.
羅吉斯分析結果的解讀(二) ▪ ln(p/1-p)=- 2.034+1.253SEX+2.058OLD ▪ 截距(β0)
=-2.034當性別為女性(0),年齡 45歲以下(0)時的估計係數 ▪ p/(1-p)= exp(-2.034)=0.131 (取指數) – 在女性且為45歲以下時有CHD的機率是沒得CHD 的機率的0.131倍
34.
羅吉斯分析結果的解讀(二) ▪ ln(p/1-p)=-2.034+1.253SEX+2.058OLD ▪ 性別(β1)
=1.253當性別為男性,年紀為45歲 以下時的估計係數 ▪ p/(1-p)= exp(1.253)=3.5 (取指數) – 45歲以下時男性得CHD的機率是是沒得CHD的機率的 3.5倍 – OLD(β2) =2.058當性別為女性,年紀為46歲以上時 的估計係數 ▪ p/(1-p)= exp(2.058)=7.83 (取指數) – 女性46歲以上時得CHD的機率是沒得CHD的機率的 7.83倍
35.
羅吉斯分析結果的解讀(二) ▪ ln(p/1-p)=- 2.034+1.253SEX+2.058OLD ▪ 當性別為男性,年紀為45歲以下時的估計係數 ▪
p/(1-p)= exp(β0+ β1)=exp(- 2.034+1.253) =0.458 – 45歲以下時得CHD的機率是男性是女性的3.5倍 ▪ OLD(β2) =2.058當性別為女性,年紀為46 歲以上時的估計係數 ▪ p/(1-p)= exp(2.058)=7.83 (取指數) – 女性46歲以上時得CHD的機率是45歲以下的7.83 倍
36.
羅吉斯分析結果的解讀(二) ▪ 男性且>46歲得CHD的機率 ▪ ln(p/1-p)=- 2.034+1.253SEX+2.058OLD ▪
p/(1-p)=exp(β1+ β2)=exp(1.253+2.058) =exp(3.311)=27.4
37.
羅吉斯分析結果的解讀(三) ▪ 當自變數有連續及類別, sex
and age ▪ -2 LL: 值愈大表示模型愈差,107.353通常並不解釋, 多用來模型互比 ▪ Cox & Snell R2(低估)及Nagelkerke R2(高估)類 似於迴歸的R2 ▪ H & L不顯著的卡方值表示資料經轉換後與線性模 型配適良好 -2 LL Pseudo R2 Hosmer & Lemeshow test Model Cox & Snell R2 Nagelkerke R2 卡方 df 顯著性 101.245 .298 .400 4.245 8 .834 Dependent variable: CHD
38.
Hosmer-Lemeshow problem ▪ Hosmer and
Lemeshow 提 出:Hosmer-Lemeshow估計程序會受 到樣本數的影響 ▪ 當樣本數大的時候,即使模型有良好的配 適,卻仍會得到顯著(P<.05)的結果 ▪ 當樣本數較小的時候,即使模型配適度不 好,仍可能得到不顯著(P>.05)的結
39.
羅吉斯分析結果的解讀(三) 模型係數的 Omnibus test step
1 卡方 df 顯著性 步驟 35.418 2 .000 區塊 35.418 2 .000 模型 35.418 2 .000 卡方值=35.418是指加了兩個自變數之後,模型卡方 值減少的值,而且是顯著的 空(截距)模型的卡方值=35.418+101.245=136.663
40.
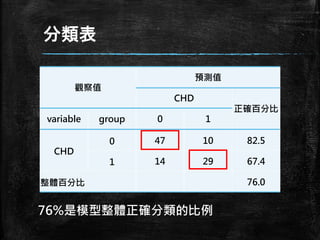
分類表 觀察值 預測值 CHD 正確百分比 variable group 0
1 CHD 0 47 10 82.5 1 14 29 67.4 整體百分比 76.0 76%是模型整體正確分類的比例
41.
敏感度 (Sensitivity) ▪ P(正確預測|事件發生) 事件發生而正確被預測到的機率 ▪
P(正確預測CHD|CHD發生) ▪ 29/(14+29) = 67.4% 觀察值 預測值 CHD 正確百分比 variable group 0 1 CHD 0 47 10 82.5 1 14 29 67.4 整體百分比 76.0
42.
明確性 (Specificity) ▪ P(正確預測|事件沒有發生) 事件沒有發生而正確被預測到沒有發生 的機率 ▪
P(正確預測沒有CHD|CHD沒有發生) ▪ 47/(47+10) = 82.5% 觀察值 預測值 CHD 正確百分比 variable group 0 1 CHD 0 47 10 82.5 1 14 29 67.4 整體百分比 76.0
43.
假陽性率(False Positive Rate) ▪
P(錯誤預測|事件發生) 事件發生而沒有被預測到事情發生的機 率 ▪ P(沒有預測到CHD|CHD發生) ▪ 10/(29+10) = 25.6% 觀察值 預測值 CHD 正確百分比 variable group 0 1 CHD 0 47 10 82.5 1 14 29 67.4 整體百分比 76.0
44.
假陰性率(False Negative Rate) ▪
P(錯誤預測|事件沒有發生) 事件沒有發生但確預測到事情會發生的 機率 ▪ P(預測到CHD|CHD沒有發生) ▪ 14/(47+14) = 23% 觀察值 預測值 CHD 正確百分比 variable group 0 1 CHD 0 47 10 82.5 1 14 29 67.4 整體百分比 76.0
45.
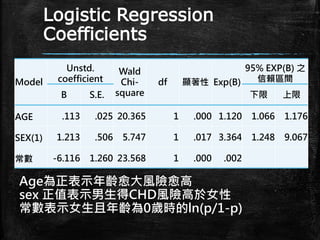
Logistic Regression Coefficients Model Unstd. coefficient Wald Chi- square df
顯著性 Exp(B) 95% EXP(B) 之 信賴區間 B S.E. 下限 上限 AGE .113 .025 20.365 1 .000 1.120 1.066 1.176 SEX(1) -1.213 .506 5.747 1 .017 .297 .110 .801 常數 -4.903 1.160 17.868 1 .000 .007 95%信心水準下,age 和 sex p<.05都是顯著的,且信 賴區間不包含1. ln(p/1-p)=-4.903-1.212sex+.113age Odds=exp(-4.903-1.212sex+.113age)
46.
Logistic Regression Coefficients Model Unstd. coefficient Wald Chi- square df 顯著性
Exp(B) 95% EXP(B) 之 信賴區間 B S.E. 下限 上限 AGE .113 .025 20.365 1 .000 1.120 1.066 1.176 SEX(1) 1.213 .506 5.747 1 .017 3.364 1.248 9.067 常數 -6.116 1.260 23.568 1 .000 .002 Age為正表示年齡愈大風險愈高 sex 正值表示男生得CHD風險高於女性 常數表示女生且年齡為0歲時的ln(p/1-p)
47.
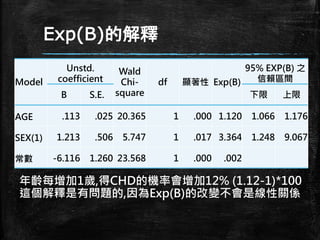
Exp(B)的解釋 Model Unstd. coefficient Wald Chi- square df 顯著性 Exp(B) 95%
EXP(B) 之 信賴區間 B S.E. 下限 上限 AGE .113 .025 20.365 1 .000 1.120 1.066 1.176 SEX(1) 1.213 .506 5.747 1 .017 3.364 1.248 9.067 常數 -6.116 1.260 23.568 1 .000 .002 年齡每增加1歲,得CHD的機率會增加12% (1.12-1)*100 這個解釋是有問題的,因為Exp(B)的改變不會是線性關係
48.
Hypothesis ▪ 本研究假設 1. 年齡愈大愈容易得CHD 2.
男性比女性容易得CHD ▪ MODEL: P(CHD) = Exp(a+b1(sex)+b2(age)) ln(p/1- p)=a+b1(sex)+b2(age)
49.
二元羅吉斯迴歸在做什麼? 1. 依變數只有兩個結果 (1=success
or 0=failure) 2. 檢定迴歸模型的配適度 3. 估計係數的顯著性 4. 估計依變數發生的機率(係數的解釋) 5. 估計迴歸係數與信賴區間 6. 結果的推論 7. 羅吉斯模型建立的應用
50.
二元羅吉斯迴歸結果的測量 1. 輸出結果的機率是由事件發生的機率 比來評估 2. 假如
P 是事件發生的機率,那麼 (1-P) 就是事件不會發生的機率 3. Odds of success = P/1-P
51.
什麼是機率(Probability)? ▪ P = 𝒐𝒖𝒕𝒄𝒐𝒎𝒆
𝒐𝒇 𝒊𝒏𝒕𝒓𝒆𝒔𝒕 𝒂𝒍𝒍 𝒑𝒐𝒔𝒔𝒊𝒃𝒍𝒆 𝒐𝒖𝒕𝒄𝒐𝒎𝒆 P = 有興趣的結果 所有可能的結果 ▪ 公正的銅版 P(正面) = 𝟏 𝟐 = . 𝟓 ▪ 公正的骰子 P(1 or 2) = 𝟐 𝟔 = 𝟏 𝟑 = .333 ▪ 一付撲克牌 P(紅心) = 𝟏𝟑 𝟓𝟐 = 𝟏 𝟒 = .25
52.
什麼是勝算(Odds)? ▪ Odds = 𝑷(𝑶𝒄𝒄𝒖𝒓𝒊𝒏𝒈) 𝑷(𝒏𝒐𝒕
𝑶𝒄𝒄𝒖𝒓𝒊𝒏𝒈) Odds = 𝑷(成功) 𝑷(失敗) = 𝑷 𝟏−𝑷 ▪ 公正的銅版 Odds(正面) = .𝟓 .𝟓 = 𝟏 ▪ 公正的骰子 Odds(1 or 2) = 𝟐 𝟒 = 𝟏 𝟐 = .5 ▪ 一付撲克牌 Odds(紅心) = 𝟏𝟑 𝟑𝟗 = 𝟏 𝟑 = .333
53.
什麼是勝算比(Odds ratio)? ▪ Odds
ratio= 𝑶𝒅𝒅𝒔𝟏 𝑶𝒅𝒅𝒔𝟐 Odds ratio= 𝑷𝟏 𝟏−𝑷𝟏 𝑷𝟎 𝟏−𝑷𝟎 ▪ Odds ratio= 𝑷𝟏 𝟏−𝑷𝟏 𝑷𝟎 𝟏−𝑷𝟎 ▪ 公正的銅版 P(正面) = 𝟏 𝟐 = . 𝟓 Odds(正面) = .𝟓 .𝟓 = 𝟏 ▪ 不公正的銅版 P(正面) = 𝟕 𝟏𝟎 = . 𝟕 Odds(正面) = .𝟕 .𝟑 = 2.333 ▪ Odds ratio= .𝟕 .𝟑 .𝟓 .𝟓 = 2.333 不公正銅版得到正面的機率是公正銅版的2.333倍
54.
羅吉斯迴歸的Odds ratio ▪ 在羅吉斯分析中的勝算比代表
“在其它變 數保持不變的情形下,自變數每改變1個 單位,應變數機率的改變”. ▪ 例如:亂編的 – 年齡與CHD的關係(有CHD/無CHD) – 假如年齡的Odds ratio為1.07 – 表示每增加1歲,得CHD的機率會增加7% – 如以增加10歲為單位,其Odds ratio為1.95, 增加20歲為單位,其Odds ratio為3.85 ▪ 這在任何年齡差距中都是成立的
55.
Sample size ▪ K為自變數 ▪
P是模型中有發生與沒發生的比例 ▪ N = 10 k / p (Peduzzi, 1996) ▪ 例如:有3個自變數,資料中有倒債人數佔 總人數的20% ▪ N = 10 x 3 / 0.20 = 150 (minimum ) ▪ 不得少於100個樣本(Long ,1997).
56.
最後提醒 ▪ No Outliers ▪
注意預測變數共線性問題。 ▪ 如有連續變數當作二維變數使用,兩預 測變數需要先mean center。 ▪ 類別的細格中不能有為0次的細格,不得 有20%的細格少於5次。 ▪ 二元羅吉斯分組儘量平均50/50。
57.
http://www.semsoeasy.com.tw/
58.
58 三星統計服務有限公司
Download