

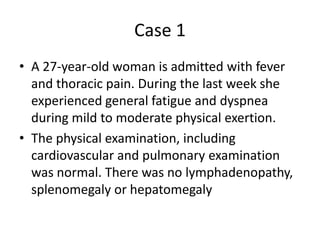
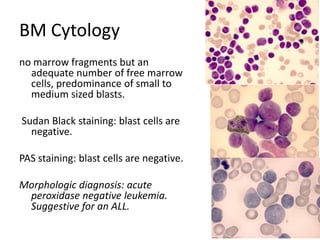

![MolecularGeneticsMultiplex RT-PCR screening for 28 fusion transcripts. Eight multiplex RT-PCR amplifications, containing each, primers for a house keeping gene as well as primers for 4 to 5 fusions transcripts are included. additional gene product: suggesting the existence of an abnormal fusion transcript. split out: abnormal band, corresponding to the fusion transcript E2A/PBX1. Conventional cytogenetics: 46, XX, del(9)(q?12q32), del(11)(p?11.2),del(11)(q22q23) [6]46, XX [11] Definitive diagnosis Pre-B-ALL with E2A/PBX1 General Remarks The fusion transcript E2A/PBX1 corresponds to a genetic translocation type t(1;19)(q23;p13). This translocation has not been found with conventional cytogenetics, probably cryptic. However, other complex cytogenetic anomalies are found on chromosome 9 and 11. pre-B-cell ALL associated with t(1;19) and/or E2A/PBX1 fusion transcript. unfavorable prognosis](https://image.slidesharecdn.com/jakobrpassweg-110630052801-phpapp02/85/LLA-2011-J-R-Passweg-Diagnosis-and-treatment-in-leukaemia-10-320.jpg)



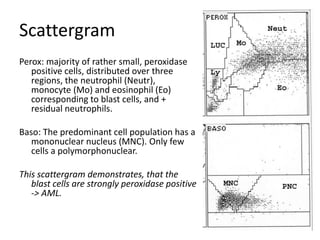
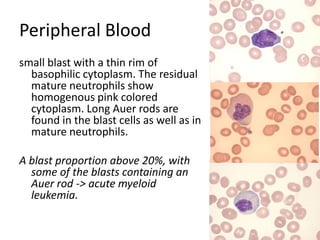
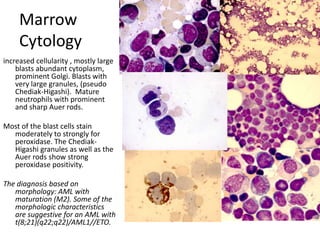
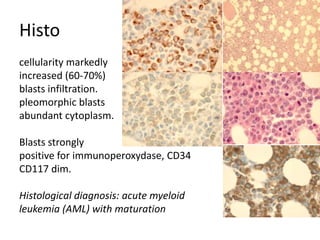
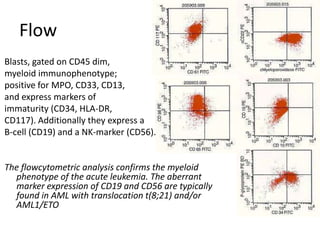
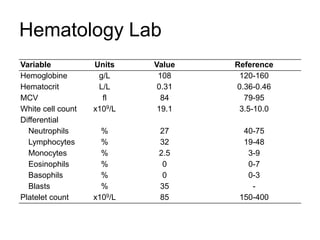
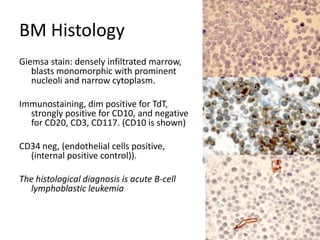
![DiagnosisMolecular genetics and Cytogenetics Using a Multiplex RT-PCR System for genotyping leukemia that screens for 28 different fusion transcripts, the blast cells are positive for the fusion transcript AML1/ETO. conventional cytogenetics, karyotype : 45, XX, -Y, t(8;21)(q22;q22) [13] Definitive Diagnosis Acute myeloid leukemia with t(8;21)(q22;q22); (AML1/ETO)](https://image.slidesharecdn.com/jakobrpassweg-110630052801-phpapp02/85/LLA-2011-J-R-Passweg-Diagnosis-and-treatment-in-leukaemia-20-320.jpg)
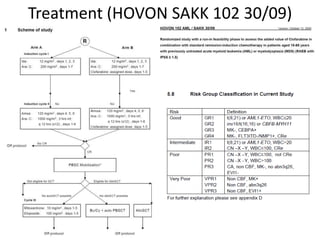
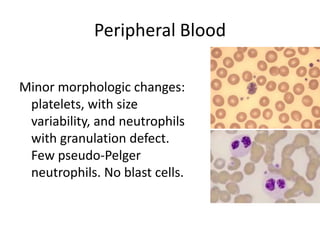
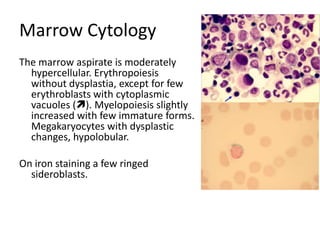
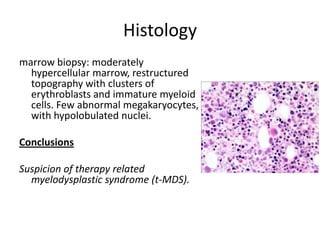

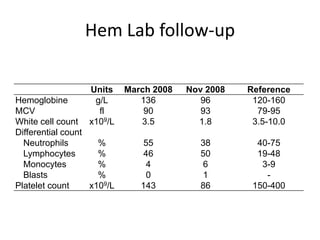

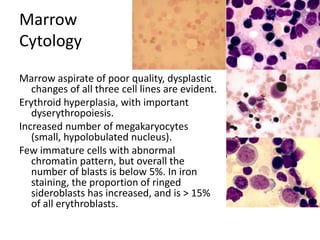
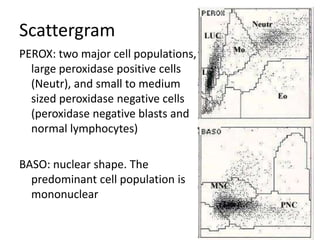
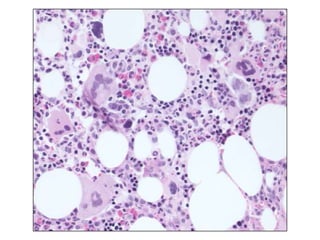
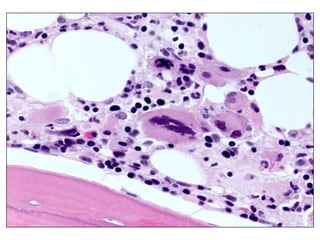
![GeneticsOnly 5 metaphases: 45, XY, add(5)(q11.2), dic(17;20)(p11.2;p112.2) [4]46, XY [1]Conclusions Therapy related myelodysplastic syndrome (t-MDS)refractory cytopenia with multilineage dysplasiawith ringed sideroblastsand cytogenetic anomalies](https://image.slidesharecdn.com/jakobrpassweg-110630052801-phpapp02/85/LLA-2011-J-R-Passweg-Diagnosis-and-treatment-in-leukaemia-32-320.jpg)
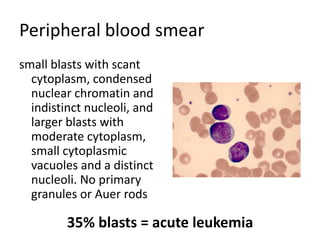
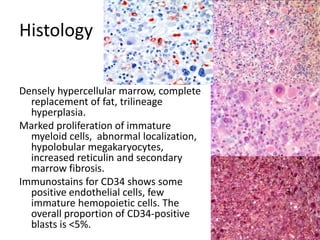
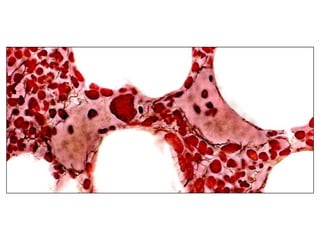
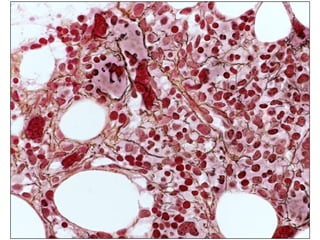
![Cytogenetics12 metaphases: 45, XY, add(5)(q11.2), dic(17;20)(p11.2;p112.2) [1]45, idem, del(7)(q22) [1]45, idem, del(7)(q22),del(12)(p11.2),-21,+r [10] Definitive diagnosis Therapy related acute myeloid Leukemia (t-AML)Alkylating agent relatedwith complex cytogenetic anomalies](https://image.slidesharecdn.com/jakobrpassweg-110630052801-phpapp02/85/LLA-2011-J-R-Passweg-Diagnosis-and-treatment-in-leukaemia-34-320.jpg)

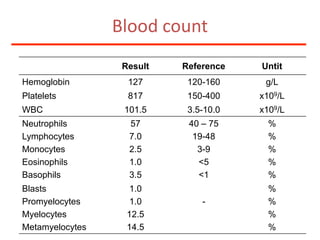
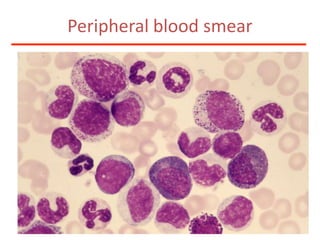




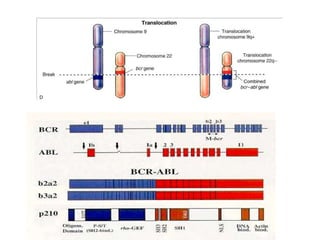

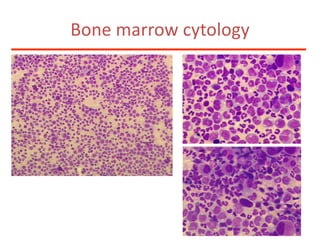
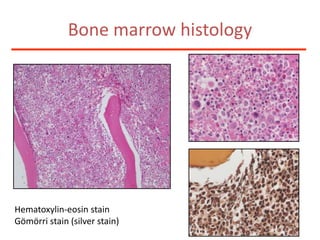
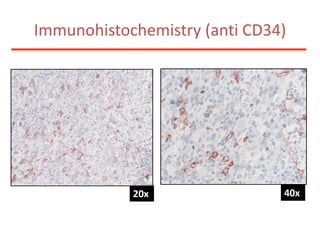

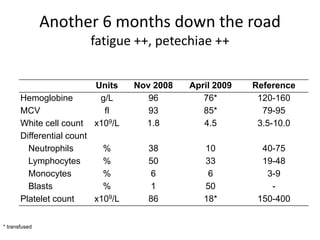
![Additional informationBonemarrowcytologyBlasts 1.5%, Basophils1%BonemarrowhistologyCD34 positive cells 2.5%Cytogenetics46,XX,t(9;22)(q34;q11)[20]](https://image.slidesharecdn.com/jakobrpassweg-110630052801-phpapp02/85/LLA-2011-J-R-Passweg-Diagnosis-and-treatment-in-leukaemia-49-320.jpg)







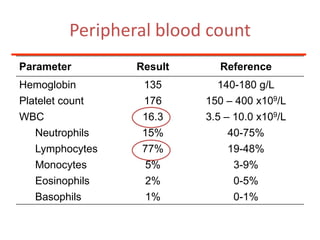
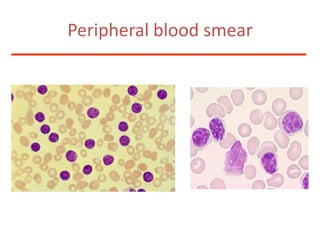
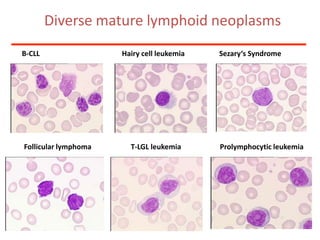


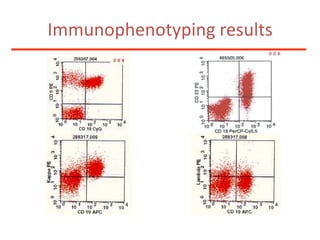
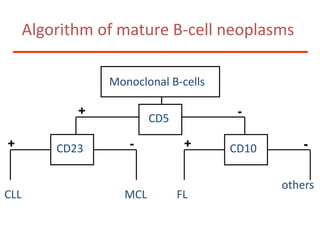
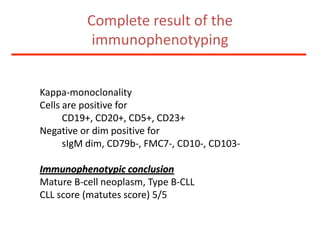


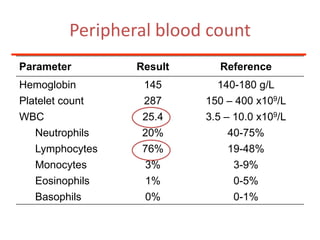
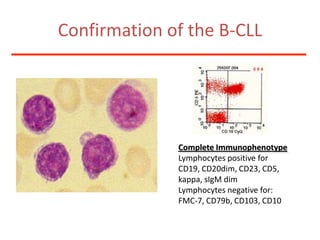


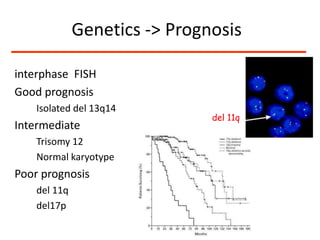




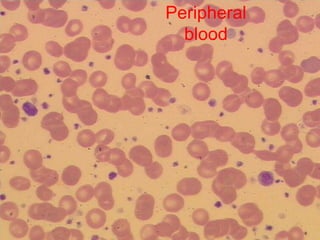
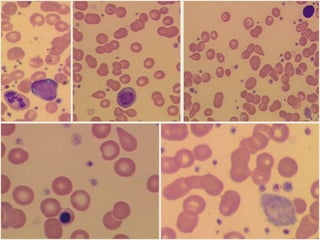

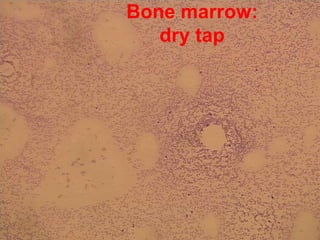
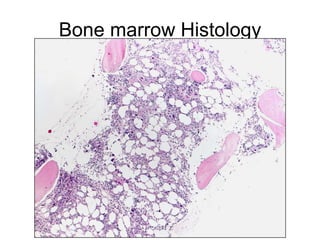
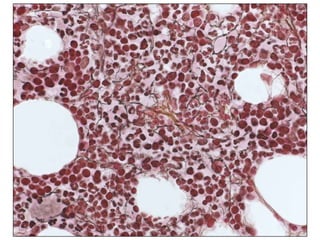










1. A 27-year-old woman was admitted with fever and chest pain and was diagnosed with acute B-cell lymphoblastic leukemia (B-ALL) with the E2A-PBX1 fusion transcript based on morphological, immunophenotypic and cytogenetic analysis. 2. A 51-year-old man presented with cough, fever and petechiae and was diagnosed with acute myeloid leukemia (AML) with maturation based on the presence of Auer rods and the AML1-ETO fusion transcript. 3. A 53-year-old man underwent follow-up 10 years after treatment for follicular lymphoma and was later diagnosed with therapy-related myelodysplastic syndrome (




































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)














![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)



