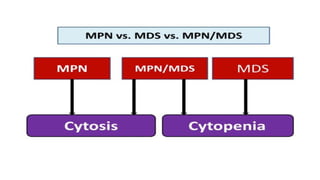
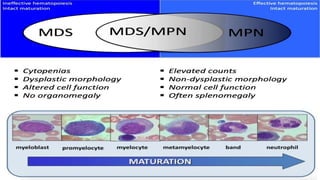
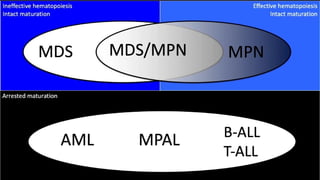
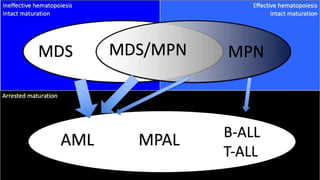
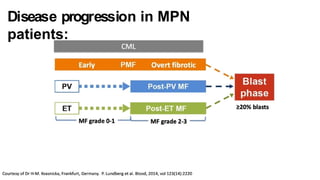
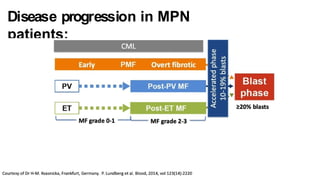
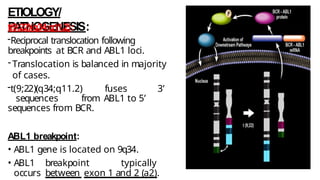
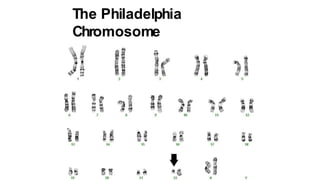
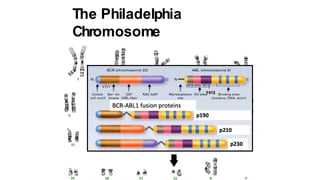
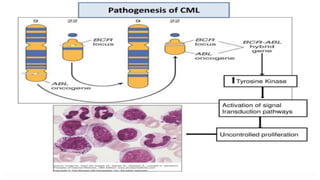
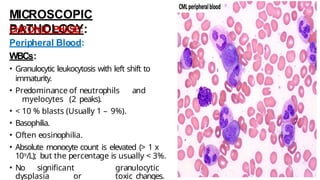
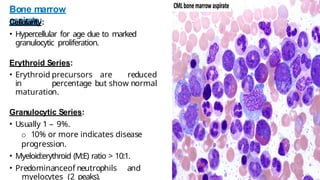
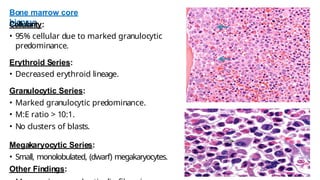
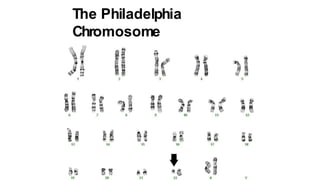
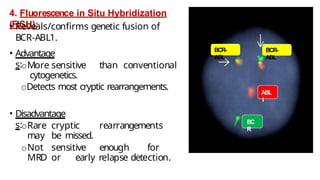
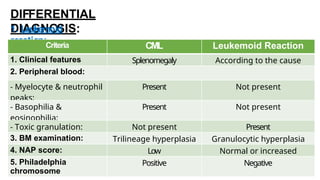
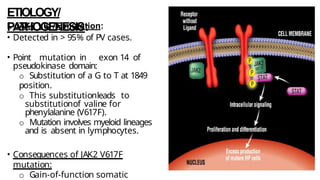
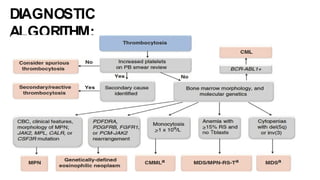
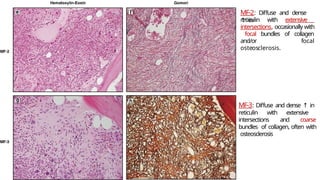
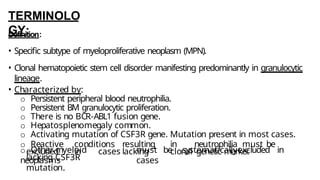
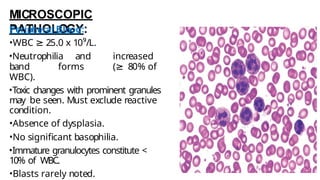
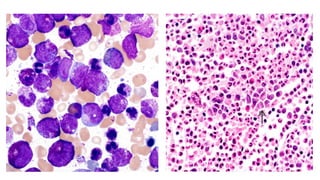
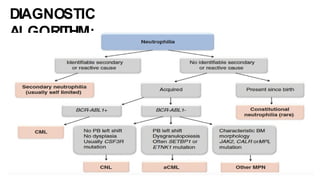
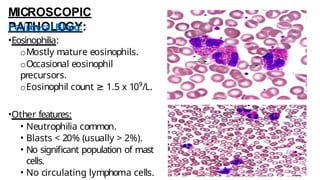
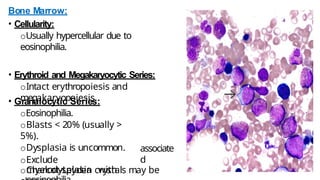
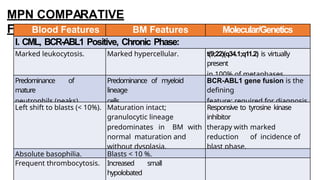
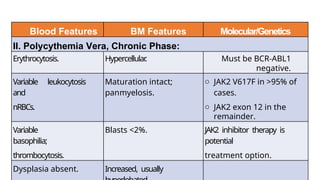
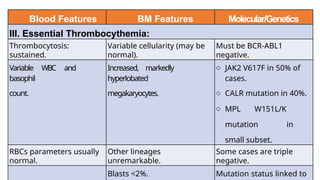
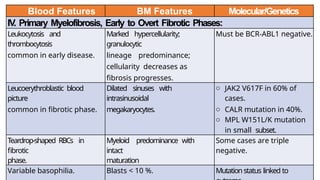
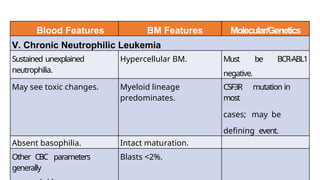
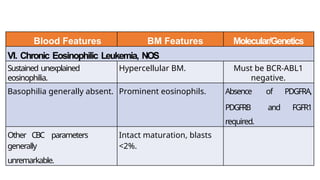
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic disorders characterized by an overproduction of blood cells, often leading to splenomegaly and hepatomegaly. The 2017 WHO classification outlines various subtypes, including chronic myeloid leukemia (CML), which is defined by the presence of the BCR-ABL1 fusion gene resulting from chromosome translocation. Disease progression in MPNs can lead to severe complications, including transformation to blast phase and bone marrow failure.









































































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)













![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)