Interstitium
• Interstitium:
– Asmall area, space, or gap:
• In the substance of an organ or tissue
– The fluid in this space is called interstitial fluid
• Comprises water and solutes,
– It drains into the lymph system
– The non-fluid part predominantly comprises of:
• Collagen fibers
– Types I, III, and V, elastin
• Glycosaminoglycans:
– Hyaluronate
– Proteoglycans that are cross-linked to form a
» Honeycomb-like reticulum.
5.
5
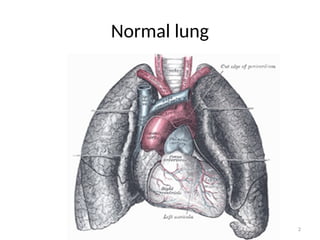
Pulmonary Interstitium
• Thespace around the alveoli:
– The interstitium is the site of gaseous exchange
– O2 leaves the lungs into the pulmonary blood vessels
– CO2 leaves the pulmonary vasculature into lungs
– It is a collection of connective tissues:
• Within the lung that includes the:
– The alveolar epithelium,
– Pulmonary capillary endothelium,
– Basement membrane,
– Peri-vascular tissues
– Peri lymphatic tissues
6.
6
ILD/DPLD
– ILD: Mayoccur when a lung injury triggers:
• An abnormal interstitial healing response
• Abnormal interstitial repair process
– The interstitium consequently becomes:
• Scarred and Thickened (fibrotic)
– The most important pathology in ILD
» Hence:
• Difficult for O2 to pass into pulmonary blood vessels
• Difficult for CO2 to pass into alveoli
7.
7
ILD/DPLDs:
– ILD/DPLD: hashigh mortality rate
• In 2013 ILD affected:
– ~ 600,000 people globally
» This resulted in > 450,000 deaths (MR ~ 75%)
8.
8
Pathology
• ILD/DPLDs:
– Havea common pathology:
• Characterized by diffuse scarring and fibrosis
– Consequently, they share features:
» Clinical
» Physiological
» Radiological
9.
9
Common Clinical features
•History:
– Common symptoms
• Cough:
– Typically: Dry, Distressing and Persistent
• Difficulty in breathing (breathlessness):
– Insidious in onset,
» But relentless in progression
• Physical examination:
• Common signs
– Bi-basal (diffuse) lower inspiratory crackles/crepitations
– Digital clubbing
10.
10
Common Lung FunctionTest features
• Lung Function Tests: reveal the following:
• Restrictive Ventilatory Defect:
– Due to decreased lung compliance
» The FEV1 and FVC are both reduced proportionally
» The FEV1 and FVC ration:
• May be normal or increased:
• Small lung volumes
• Reduced Gas Transfer
– All these being consequences of relentless:
» Fibrosis and scarification
11.
11
Common radiological features
•Typical radiographic features:
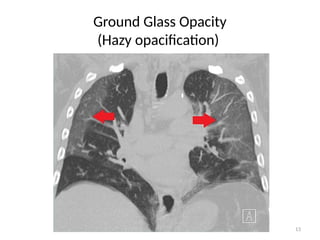
– Early stages of disease:
• Ground Glass Opacity (GGO)
– An area of hazy opacification (on X ray) or
– Increased attenuation (on CT scan)
» Due to air displacement by: fluid, airway collapse, and fibrosis
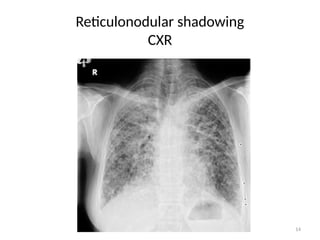
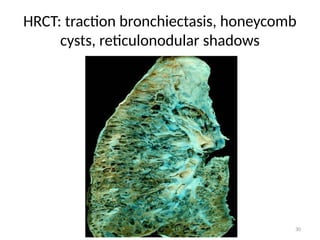
• Reticulonodular shadowing:
– There is an overlap of reticular shadows with nodular shadows
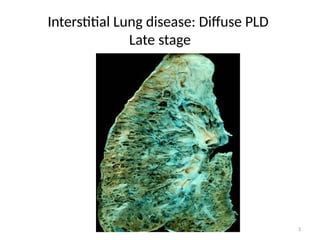
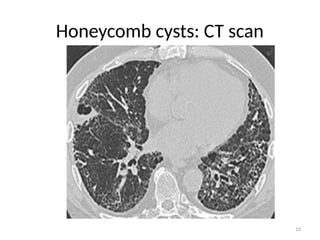
– Late stages:
• Honey combed cysts
– Due to widespread fibrosis:
» Small cystic spaces form with irregularly thickened walls
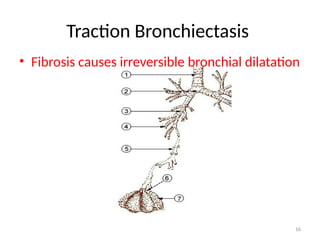
• Traction bronchiectasis
» Scar tissue applying traction force on bronchi, hence dilation
12.
12
Traction bronchiectasis
• Asubtype of bronchiectasis where there is:
– irreversible dilatation of bronchi and bronchioles
• Within areas of pulmonary fibrosis
– The bronchus is pulled apart:
• By the traction of surrounding lung fibrosis.
– There may be a preference for the upper lobes
• Where there is less supporting cartilage
23
IDIOPATHIC INTERSTITIAL PNEUMONIAS
•A major subgroup of DPLDs
• Contains several subtypes
• Grouped together due to their unknown etiology
• Distinguished by characteristic histologic pattern on
tissue biopsy
– Most important of these is:
• Idiopathic Pulmonary Fibrosis
24.
24
IDIOPATHIC PULMONARY FIBROSIS
–A progressive Fibrosing Interstitial Pneumonia:
• Of unknown etiology occurring in adults.
– Histological and radiological features constitute the term:
» “Usual” Interstitial Pneumonia (UIP)
• Histological features suggest the cause as being:
– Repeated focal trauma to alveolar epithelium:
» Which is consistent with an autoimmune process.
25.
25
Clinical features:
• Elderlypresentation:
– Rare before 50yrs
– May be an incidental discovery:
• Following CT scanning in asymptomatic individual.
– Typical presentation:
• Dry cough
• Difficulty in breathing (Breathlessness):
– Insidious onset,
– relentless in progression
• No constitutional symptoms: (NO fever, NO sweating,)
– Clinical/physical findings:
• Finger clubbing,
• Bi-basal fine late inspiratory crackles
27
– Biochemistry
• SerumCa 2+
:
– Elevated in sarcoidosis
• Serum Lactate Dehydrogenase:
– High in acute alveolitis
• Serum ACE:
– Indicator of disease activity in sarcoidosis:
» Non specific
– Autoimmune/serology screen:
• Autoantibodies
– Lung disease may precede connective tissue disease
28.
28
• B: Radiology:
–CXR:
• Usually the first test to detect ILD
• But can be normal in up to 10% of patients
– Especially in early disease.
• Radiological Features:
– Small lung volumes
» Shrunken lungs
– Reticulonodular shadowing:
» Seen in lower lobe and sub-pleural area bilaterally
29.
29
HRCT
– HRCT:
• Ground-Glass Opacity/GGO
– Area of increased attenuation in the lung
• Honey comb cysts
• Traction bronchiectasis:
• Reticulonodular shadows:
– Patchy predominantly peripheral, basal and sub-pleural
31
Lung Function Tests
•Restrictive ventilatory defect:
– Spirometry:
• Forced expiratory volume in the 1st
sec
• Forced vital capacity
– Calculate ratio: FEV1/FVC
» Normally reduced,
• Could be high or normal in advanced disease
– Reduced lung volumes
• Proportional reduction of FEV in 1st
sec and FVC
– Impaired gas transfer.
• Oxygen out of alveoli from pulmonary capillaries
• Carbon dioxide from pulmonary capillaries into alveoli
32.
32
• Bronchoscopy:
– Seldomnecessary
• Unless there is a high chance of infection or malignancy
– Broncho-alveolar lavage:
• Broncho-alveolar fluid cell count, may point to:
– Sarcoid pneumonitis
– Drug induced pneumonitis
– Pulmonary eosinophilia
– Hypersensitivity pneumonitis
– Cryptogenic organizing pneumonitis
– Infection
33.
33
• Trans-bronchial lungbiopsy:
– Useful in
• Sarcoidosis, malignancy, infection
• Bronchial biopsy:
– Useful in
• Sarcoidosis
• Video-assisted thorascopic lung biopsy:
– Selected cases:
• For pathological classification:
– Asbestos in asbestosis,
– Silica in occupational fibrosing lung disease
34.
34
Management
• Treatment:
– Generallydifficult
– ILD is not a single disease
• It involves many pathological processes.
– Hence treatment is different for each disease.
– If a specific occupational cause is found:
• Avoid that environment.
– If a drug is suspected:
• Discontinue the drug
35.
35
Drug treatment
• Combinationtreatment:
– Drugs aiming at reduction of inflammation and fibrosis
• Prednisolone
• Azathioprine
• N-acetylcysteine,
– All Had shown promise at some points:
» Not supported by large studies
– Other drugs:
• Colchicine,
• Interferone –γ 1b,
• Etanercept,
• Bosentan:
– Disappointing results
36.
36
Lung transplant
• Lungtransplantation:
– An option if the ILD progresses
• Despite therapy in appropriately selected patients:
– With no contraindications
37.
37
• Look forand treat:
• GERD (in patients with dyspepsia)
• Pulmonary hypertension
• Give Oxygen:
• In breathless individuals,
38.
38
PROGNOSIS:
• Natural history:
–The prognosis is poor
– There is usually a steady decline
– In some patients:
• There are exacerbations with acute worsening of
– Breathlessness
– Disturbed gas exchange
– New GGO or consolidation on HRCT
– With advanced disease:
• Central cyanosis, RHF
– Median survival:
• 3 years, may vary widely
– Serial LFTs:
• Provide useful prognostic information
– IPF is associated with lung cancer.
39.
39
References
• Recommended textbooks
– Davidson’s Principles & Practice of Medicine
• 22nd
Ed and above
– Kumar & Clark’s Clinical Medicine
• 17th
Ed and above
















































![Interstitial Lung Diseases [ILD] Approach to Management](https://cdn.slidesharecdn.com/ss_thumbnails/interstitiallungdiseases-arunvasireddy-19october2015-seminar-171016041856-thumbnail.jpg?width=640&height=640&fit=bounds)





































