Download to read offline



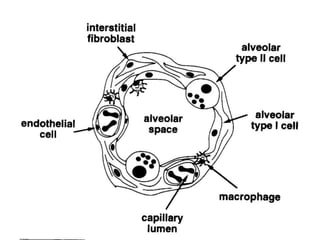





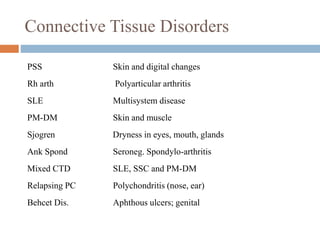




























Interstitial lung disease (ILD) encompasses over 250 different diseases affecting the lung interstitium, characterized by inflammation and fibrosis. The document discusses the causes, classifications, diagnosis, and treatment of ILD, highlighting idiopathic pulmonary fibrosis as a prevalent form with a poor prognosis. Diagnostic approaches include clinical features, imaging, pulmonary function tests, and biopsy, while treatments aim to alleviate symptoms, slow progression, and improve quality of life.














![Interstitial Lung Diseases [ILD] Approach to Management](https://cdn.slidesharecdn.com/ss_thumbnails/interstitiallungdiseases-arunvasireddy-19october2015-seminar-171016041856-thumbnail.jpg?width=640&height=640&fit=bounds)




















































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)