Downloaded 540 times


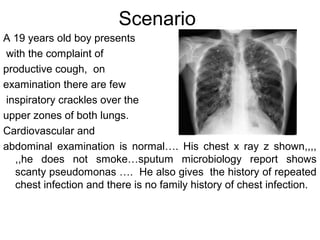
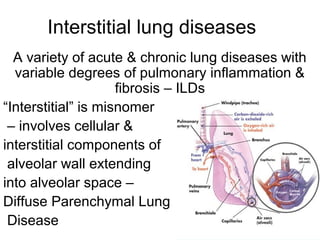
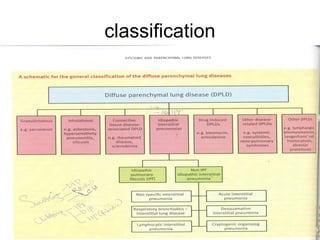
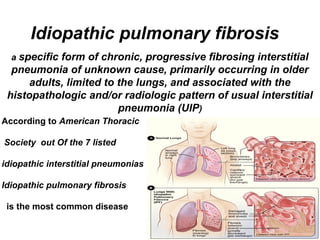


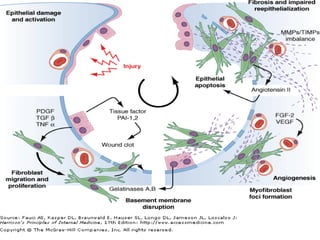


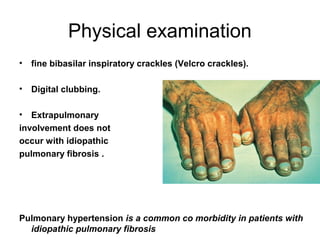






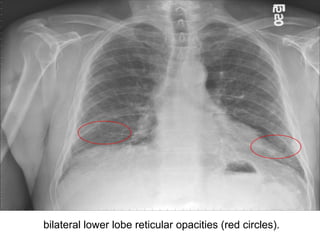

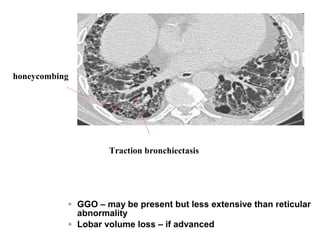
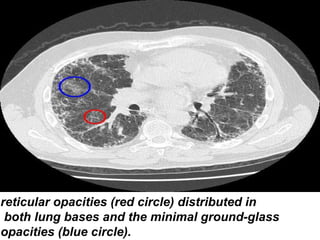





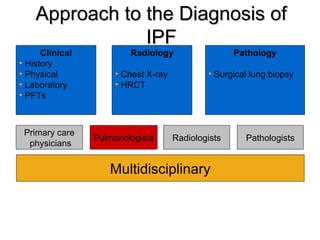








1) The patient presents with a history of recurrent chest infections and inspiratory crackles on examination. Imaging and pulmonary function tests are required to diagnose interstitial lung disease. 2) Idiopathic pulmonary fibrosis is a chronic, progressive form of interstitial lung disease of unknown cause characterized by fibrosis of the lungs. It carries a poor prognosis with median survival of 3 years. 3) Diagnosis requires ruling out other causes through history, imaging showing reticular opacities and honeycombing, and lung biopsy if imaging is not definitive. Treatment focuses on managing complications, vaccination, oxygen therapy and consideration of lung transplantation in advanced cases.























































































