The document presents a seminar by Nathalie Villa-Vialaneix on integrating Tara Oceans datasets through unsupervised multiple kernel learning. It outlines the significance of metagenomic data, the methodology for analysis using kernel methods, and discusses the Tara Oceans project, which collected extensive data on oceanic plankton diversity. The aim is to develop a framework for integrating various metagenomic data to enhance understanding of ecological interactions.




![What are metagenomic data?
Source: [Sommer et al., 2010]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 4/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-4-320.jpg)
![What are metagenomic data?
Source: [Sommer et al., 2010]
abundance data sparse
n × p-matrices with count data
of samples in rows and
descriptors (species, OTUs,
KEGG groups, k-mer, ...) in
columns. Generally p n.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 4/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-5-320.jpg)
![What are metagenomic data?
Source: [Sommer et al., 2010]
abundance data sparse
n × p-matrices with count data
of samples in rows and
descriptors (species, OTUs,
KEGG groups, k-mer, ...) in
columns. Generally p n.
philogenetic tree (evolution
history between species,
OTUs...). One tree with p leaves
built from the sequences
collected in the n samples.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 4/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-6-320.jpg)









![TARA Oceans datasets
Science (May 2015) - Studies on:
eukaryotic plankton diversity
[de Vargas et al., 2015],
ocean viral communities
[Brum et al., 2015],
global plankton interactome
[Lima-Mendez et al., 2015],
global ocean microbiome
[Sunagawa et al., 2015],
. . . .
→ datasets from different types and
different sources analyzed separately.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 10/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-16-320.jpg)

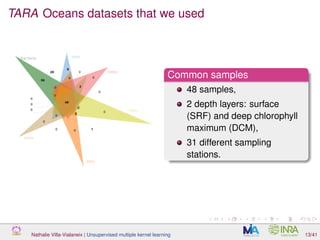
![TARA Oceans datasets that we used
[Sunagawa et al., 2015]
Datasets used
environmental dataset: 22 numeric features (temperature, salinity, . . . ).
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 12/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-18-320.jpg)
![TARA Oceans datasets that we used
[Sunagawa et al., 2015]
Datasets used
environmental dataset: 22 numeric features (temperature, salinity, . . . ).
bacteria phylogenomic tree: computed from ∼ 35,000 OTUs.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 12/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-19-320.jpg)
![TARA Oceans datasets that we used
[Sunagawa et al., 2015]
Datasets used
environmental dataset: 22 numeric features (temperature, salinity, . . . ).
bacteria phylogenomic tree: computed from ∼ 35,000 OTUs.
bacteria functional composition: ∼ 63,000 KEGG orthologous groups.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 12/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-20-320.jpg)
![TARA Oceans datasets that we used
[de Vargas et al., 2015]
Datasets used
environmental dataset: 22 numeric features (temperature, salinity, . . . ).
bacteria phylogenomic tree: computed from ∼ 35,000 OTUs.
bacteria functional composition: ∼ 63,000 KEGG orthologous groups.
eukaryotic plankton composition splited into 4 groups pico (0.8 − 5µm),
nano (5 − 20µm), micro (20 − 180µm) and meso (180 − 2000µm).
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 12/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-21-320.jpg)
![TARA Oceans datasets that we used
[Brum et al., 2015]
Datasets used
environmental dataset: 22 numeric features (temperature, salinity, . . . ).
bacteria phylogenomic tree: computed from ∼ 35,000 OTUs.
bacteria functional composition: ∼ 63,000 KEGG orthologous groups.
eukaryotic plankton composition splited into 4 groups pico (0.8 − 5µm),
nano (5 − 20µm), micro (20 − 180µm) and meso (180 − 2000µm).
virus composition: ∼ 867 virus clusters based on shared gene content.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 12/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-22-320.jpg)


![Kernel methods
Kernel viewed as the dot product in an implicit Hilbert space
K : X × X → R st: K(xi, xj) = K(xj, xi) and ∀ m ∈ N, ∀x1, ..., xm ∈ X,
∀ α1, ..., αm ∈ R, m
i,j=1 αiαjK(xi, xj) ≥ 0.
⇒ [Aronszajn, 1950]
∃!(H, ., . ), φ : X → H st: K(xi, xj) = φ(xi), φ(xj)
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 15/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-26-320.jpg)
![Exploratory analysis with kernels
A well know example: kernel PCA [Schölkopf et al., 1998]
PCA analysis performed in the feature space induced by the kernel K.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 16/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-27-320.jpg)
![Exploratory analysis with kernels
A well know example: kernel PCA [Schölkopf et al., 1998]
PCA analysis performed in the feature space induced by the kernel K.
In practice:
K is centered: K ← K − 1
N KIN + 1
N2 IN
KIN;
K-PCA is performed by the eigen-decomposition of (centered) K
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 16/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-28-320.jpg)
![Exploratory analysis with kernels
A well know example: kernel PCA [Schölkopf et al., 1998]
PCA analysis performed in the feature space induced by the kernel K.
In practice:
K is centered: K ← K − 1
N KIN + 1
N2 IN
KIN;
K-PCA is performed by the eigen-decomposition of (centered) K
If (αk )k=1,...,N ∈ RN
and (λk )k=1,...,N are the eigenvectors and eigenvalues,
PC axes are:
ak =
N
i=1
αkiφ(xi)
and ak = (aki)i=1,...,n are orthonormal in the feature space induced by the
kernel:
∀ k, k , ak , ak = αk Kαk = δkk with δkk =
0 if k k
1 otherwise
.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 16/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-29-320.jpg)
![Exploratory analysis with kernels
A well know example: kernel PCA [Schölkopf et al., 1998]
PCA analysis performed in the feature space induced by the kernel K.
In practice:
K is centered: K ← K − 1
N KIN + 1
N2 IN
KIN;
K-PCA is performed by the eigen-decomposition of (centered) K
Coordinate of the projection of the observations (φ(xi))i:
ak , φ(xi) =
n
j=1
αkjKji = Ki.αk = λk αki,
where Ki. is the i-th row of K.
No representation for the variables (no real variables...).
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 16/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-30-320.jpg)
![Exploratory analysis with kernels
A well know example: kernel PCA [Schölkopf et al., 1998]
PCA analysis performed in the feature space induced by the kernel K.
In practice:
K is centered: K ← K − 1
N KIN + 1
N2 IN
KIN;
K-PCA is performed by the eigen-decomposition of (centered) K
Other unsupervised kernel methods: kernel SOM
[Olteanu and Villa-Vialaneix, 2015, Mariette et al., 2017]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 16/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-31-320.jpg)
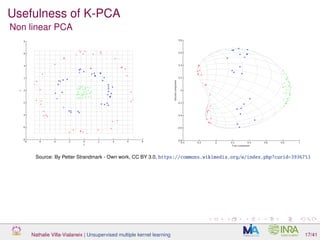
![Usefulness of K-PCA
[Mariette et al., 2017] K-PCA for non numeric datasets - here a
quantitative time series: job trajectories after graduation from the French
survey “Generation 98” [Cottrell and Letrémy, 2005]
color is the mode of the trajectories
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 17/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-33-320.jpg)
![From multiple dissimilarities to multiple kernels
1 several (non Euclidean) dissimilarities D1
, . . . , DM
, transformed into
similarities with [Lee and Verleysen, 2007]:
Km
(xi, xj) = −
1
2
Dm
(xi, xj) −
2
N
N
k=1
Dm
(xi, xk ) +
1
N2
N
k, k =1
Dm
(xk , xk )
2 if non positive, clipping or flipping (removing the negative part of the
eigenvalues decomposition or taking its opposite) produce kernels
[Chen et al., 2009].
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 18/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-34-320.jpg)

![From multiple kernels to an integrated kernel
How to combine multiple kernels?
naive approach: K∗ = 1
M m Km
supervised framework: K∗ = m βmKm
with βm ≥ 0 and m βm = 1
with βm chosen so as to minimize the prediction error
[Gönen and Alpaydin, 2011]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 19/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-36-320.jpg)
![From multiple kernels to an integrated kernel
How to combine multiple kernels?
naive approach: K∗ = 1
M m Km
supervised framework: K∗ = m βmKm
with βm ≥ 0 and m βm = 1
with βm chosen so as to minimize the prediction error
[Gönen and Alpaydin, 2011]
unsupervised framework but input space is Rd
[Zhuang et al., 2011]
K∗ = m βmKm
with βm ≥ 0 and m βm = 1 with βm chosen so as to
minimize the distortion between all training data ij K∗
(xi, xj) xi − xj
2
;
AND minimize the approximation of the original data by the kernel
embedding i xi − j K∗
(xi, xj)xj
2
.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 19/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-37-320.jpg)
![From multiple kernels to an integrated kernel
How to combine multiple kernels?
naive approach: K∗ = 1
M m Km
supervised framework: K∗ = m βmKm
with βm ≥ 0 and m βm = 1
with βm chosen so as to minimize the prediction error
[Gönen and Alpaydin, 2011]
unsupervised framework but input space is Rd
[Zhuang et al., 2011]
K∗ = m βmKm
with βm ≥ 0 and m βm = 1 with βm chosen so as to
minimize the distortion between all training data ij K∗
(xi, xj) xi − xj
2
;
AND minimize the approximation of the original data by the kernel
embedding i xi − j K∗
(xi, xj)xj
2
.
Our proposal: 2 UMKL frameworks which do not require data to have
values in Rd
.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 19/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-38-320.jpg)
![STATIS like framework
[L’Hermier des Plantes, 1976, Lavit et al., 1994]
Similarities between kernels:
Cmm =
Km
, Km
F
Km
F Km
F
=
Trace(Km
Km
)
Trace((Km)2)Trace((Km )2)
.
(Cmm is an extension of the RV-coefficient [Robert and Escoufier, 1976] to
the kernel framework)
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 20/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-39-320.jpg)
![STATIS like framework
[L’Hermier des Plantes, 1976, Lavit et al., 1994]
Similarities between kernels:
Cmm =
Km
, Km
F
Km
F Km
F
=
Trace(Km
Km
)
Trace((Km)2)Trace((Km )2)
.
(Cmm is an extension of the RV-coefficient [Robert and Escoufier, 1976] to
the kernel framework)
maximize
M
m=1
K∗
(v),
Km
Km
F F
= v Cv
for K∗
(v) =
M
m=1
vmKm
and v ∈ RM
such that v 2 = 1.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 20/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-40-320.jpg)
![STATIS like framework
[L’Hermier des Plantes, 1976, Lavit et al., 1994]
Similarities between kernels:
Cmm =
Km
, Km
F
Km
F Km
F
=
Trace(Km
Km
)
Trace((Km)2)Trace((Km )2)
.
(Cmm is an extension of the RV-coefficient [Robert and Escoufier, 1976] to
the kernel framework)
maximize
M
m=1
K∗
(v),
Km
Km
F F
= v Cv
for K∗
(v) =
M
m=1
vmKm
and v ∈ RM
such that v 2 = 1.
Solution: first eigenvector of C ⇒ Set β = v
M
m=1 vm
(consensual kernel).
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 20/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-41-320.jpg)
![A kernel preserving the original topology of the data I
From an idea similar to that of [Lin et al., 2010], find a kernel such that the
local geometry of the data in the feature space is similar to that of the
original data.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 21/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-42-320.jpg)
![A kernel preserving the original topology of the data I
From an idea similar to that of [Lin et al., 2010], find a kernel such that the
local geometry of the data in the feature space is similar to that of the
original data.
Proxy of the local geometry
Km
−→ Gm
k
k−nearest neighbors graph
−→ Am
k
adjacency matrix
⇒ W = m I{Am
k
>0} or W = m Am
k
Adjacency matrix image from: By S. Mohammad H. Oloomi, CC BY-SA 3.0,
https://commons.wikimedia.org/w/index.php?curid=35313532
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 21/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-43-320.jpg)
![A kernel preserving the original topology of the data I
From an idea similar to that of [Lin et al., 2010], find a kernel such that the
local geometry of the data in the feature space is similar to that of the
original data.
Proxy of the local geometry
Km
−→ Gm
k
k−nearest neighbors graph
−→ Am
k
adjacency matrix
⇒ W = m I{Am
k
>0} or W = m Am
k
Feature space geometry measured by
∆i(β) = φ∗
β(xi),
φ∗
β(x1)
...
φ∗
β(xN)
=
K∗
β (xi, x1)
...
K∗
β (xi, xN)
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 21/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-44-320.jpg)









![A proposal to improve interpretability of K-PCA in our
framework
Issue: How to assess the importance of a given species in the K-PCA?
our datasets are either numeric (environmental) or are built from a
n × p count matrix
⇒ for a given species, randomly permute counts and re-do the
analysis (kernel computation - with the same optimized weights - and
K-PCA)
the influence of a given species in a given dataset on a given PC
subspace is accessed by computing the Crone-Crosby distance
between these two PCA subspaces [Crone and Crosby, 1995] (∼
Frobenius norm between the projectors)
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 24/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-54-320.jpg)



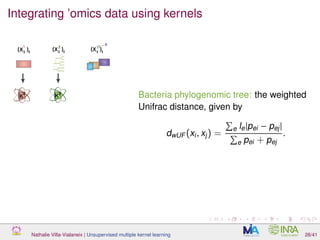


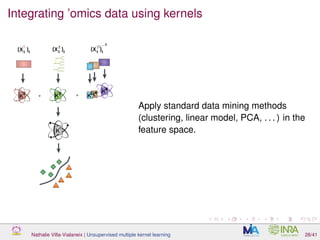
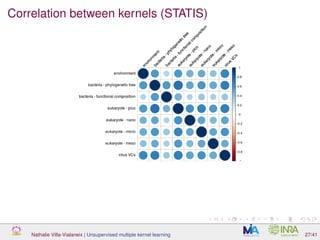
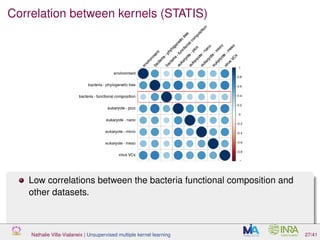
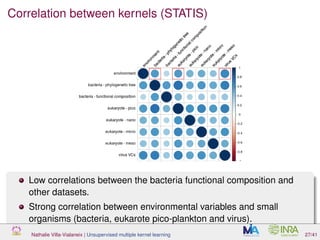
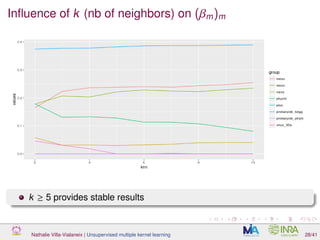
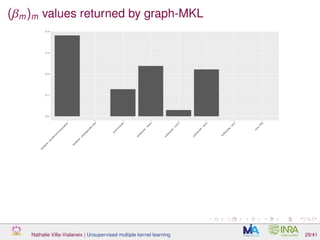
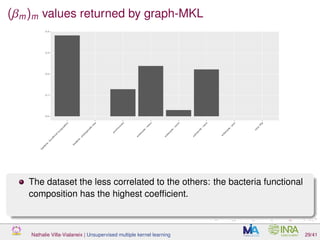
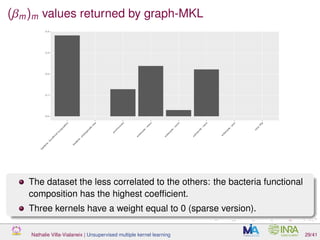
![Proof of concept: using [Sunagawa et al., 2015]
Datasets
139 samples, 3 layers (SRF, DCM and MES)
kernels: phychem, pro-OTUs and pro-OGs
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 30/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-69-320.jpg)
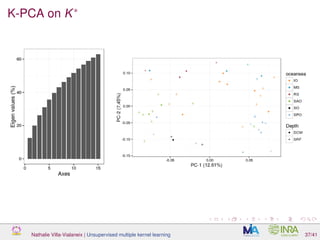
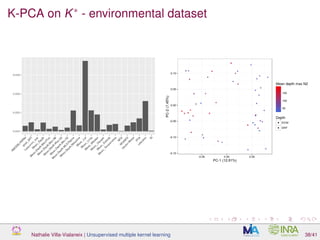
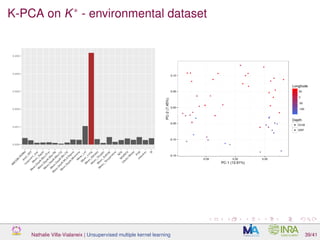
![Proof of concept: using [Sunagawa et al., 2015]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 31/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-70-320.jpg)
![Proof of concept: using [Sunagawa et al., 2015]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 32/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-71-320.jpg)
![Proof of concept: using [Sunagawa et al., 2015]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 33/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-72-320.jpg)
![Proof of concept: using [Sunagawa et al., 2015]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 34/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-73-320.jpg)
![Proof of concept: using [Sunagawa et al., 2015]
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 35/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-74-320.jpg)
![Proof of concept: using [Sunagawa et al., 2015]
Proteobacteria (clade SAR11 (Alphaproteobacteria) and SAR86)
dominate the sampled areas of the ocean in term of relative
abundance and taxonomic richness.
Nathalie Villa-Vialaneix | Unsupervised multiple kernel learning 36/41](https://image.slidesharecdn.com/slidesnice2017-01-24-170124154221/85/Integrating-Tara-Oceans-datasets-using-unsupervised-multiple-kernel-learning-75-320.jpg)