





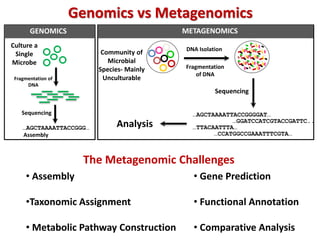


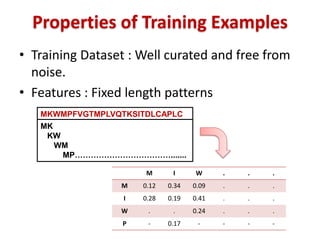


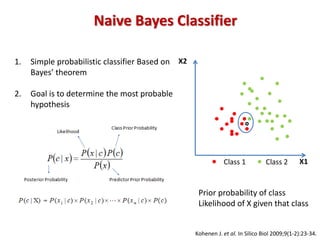
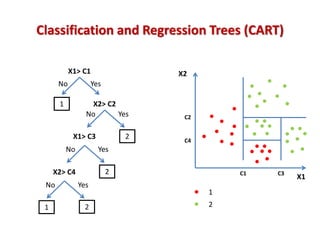
![Algorithm: Word sizes between 6 and 9 bases
Word-specific priors: Pi = [n(wi) + 0.5]/(N +1)]
Genus-specific conditional probabilities: P(wi|G) = [m(wi) + Pi]/(M + 1)
Naive Bayesian assignment: P(G|S) = P(S|G) * P(G)/P(S)
Bootstrap confidence estimation: For each query sequence
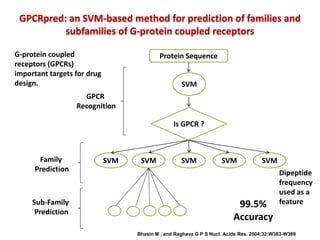
Naive Bayesian Classifier for Rapid Assignment of
rRNA Sequences into New Bacterial Taxonomy
Qiong Wang et. al., Appl Environ Microbiol, 2007
AUGCGUCAGCUCGAUCGAUCUA
AUGCGUCA
UGCGUCAG
GCGUCAGC
CGUCAGCU](https://image.slidesharecdn.com/machinelearning-150304000629-conversion-gate01/85/Machine-Learning-26-320.jpg)
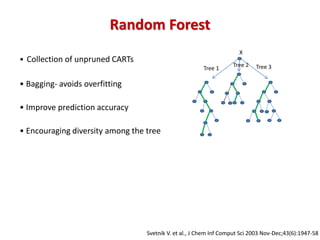

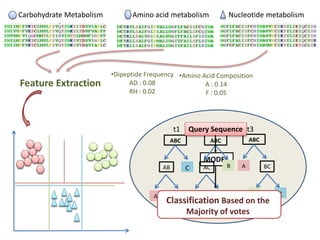

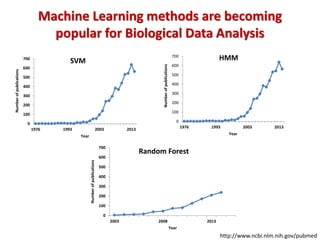









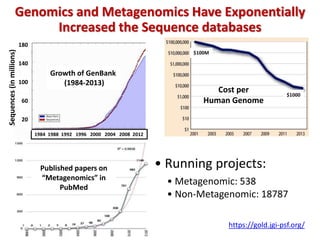
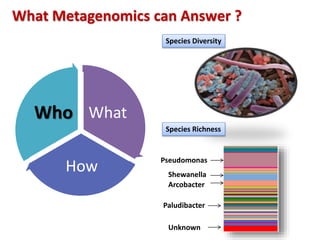
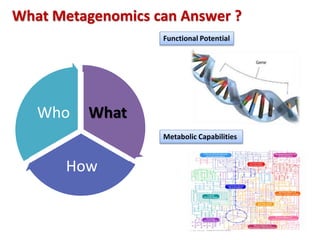
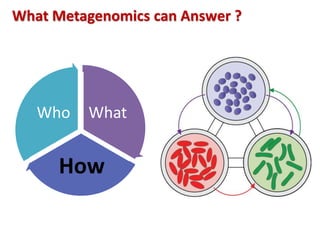
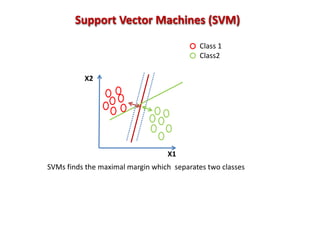
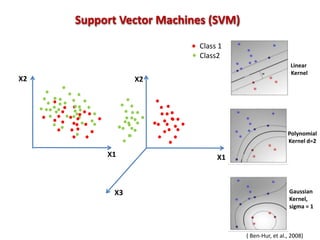
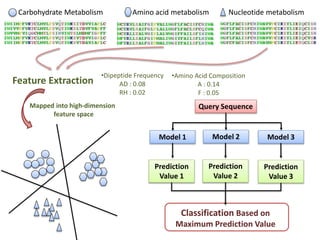
The document discusses the use of machine learning approaches for analyzing high-throughput biological data, specifically in genomics and metagenomics. It covers various machine learning techniques such as support vector machines, hidden Markov models, naive Bayes, and random forests, highlighting their applications and advantages in addressing challenges faced in conventional data analysis methods. Additionally, it outlines the historical context of genomics and future directions for machine learning applications in this field.


![[2013.10.29] albertsen genomics metagenomics](https://cdn.slidesharecdn.com/ss_thumbnails/2013-131029070115-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



































![[DSC Adria 23] Enes Deumic application of ai in genomics.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/02enesdeumicapplicationofaiingenomics-230530183921-d488ad4b-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DSC Europe 23][DigiHealth] Vesna Pajic - Machine Learning Techniques for omi...](https://cdn.slidesharecdn.com/ss_thumbnails/dxgiw6wysauhxm3dnays-vesna-pajic-machine-learning-techniques-for-omics-data-analysis-231130112724-c1268146-thumbnail.jpg?width=640&height=640&fit=bounds)


























