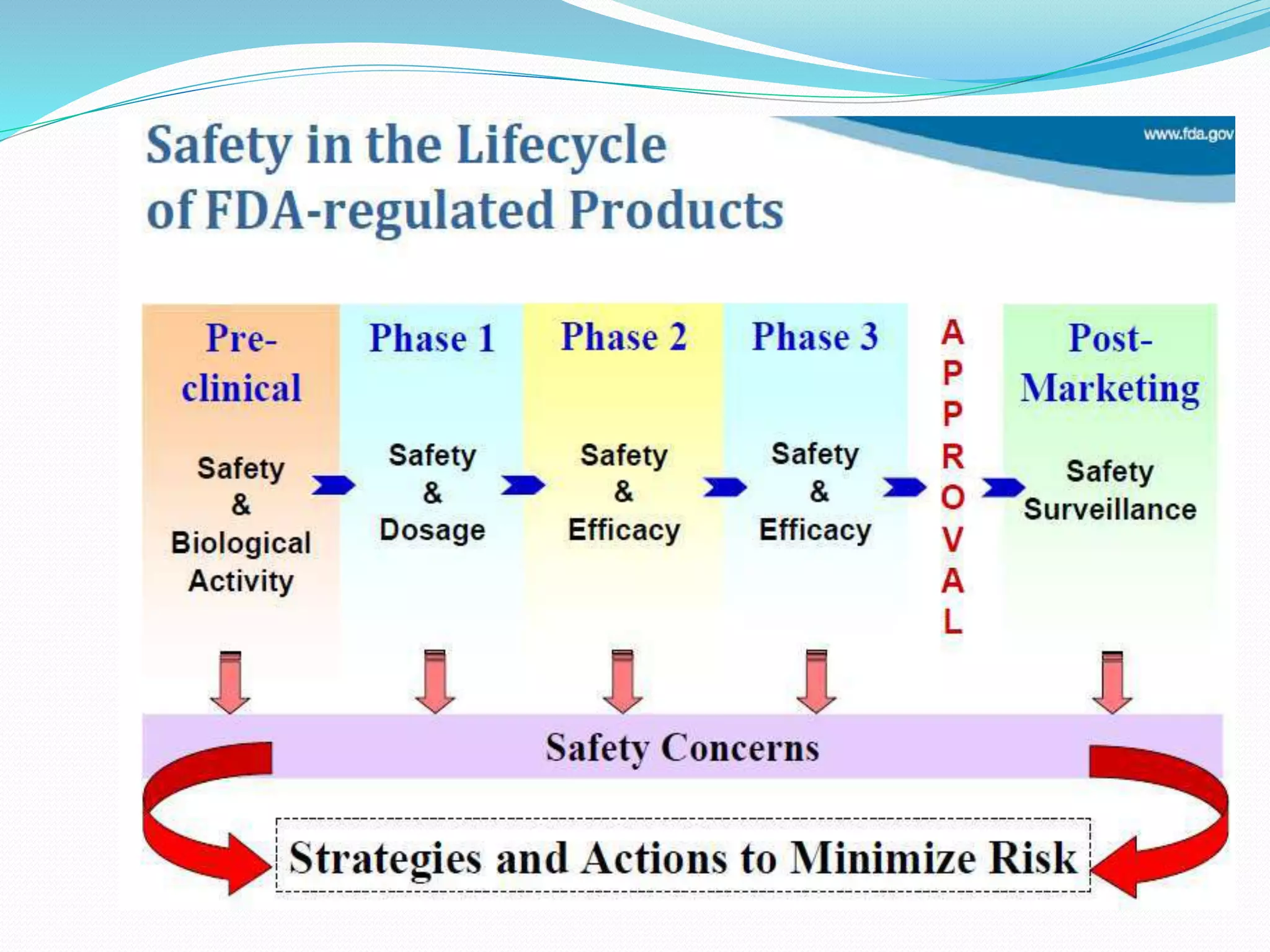
This document discusses post-marketing surveillance (PMS), which refers to monitoring drugs after they have been approved for public use. PMS is important because clinical trials have limitations in terms of patient population size, duration, and ability to detect rare or long-term effects. Sources of PMS information include spontaneous reporting, studies using medical databases, and manufacturer monitoring. Common PMS methods include spontaneous reporting of adverse drug reactions, cohort studies, and case control studies. PMS can help identify new safety issues and provide more information on drug risks and benefits in diverse patient populations.






![POST-MARKETING SURVEILLANCE
No fixed duration/Patient population
Starts immediately after marketing
Report all ADRs
Help to detect
Rare ADRs
Drug interaction
Also new uses for drugs[sometimes called
Phase V]](https://image.slidesharecdn.com/pms-171110044659/75/PMS-post-marketing-surveillance-8-2048.jpg)









































![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)


















































