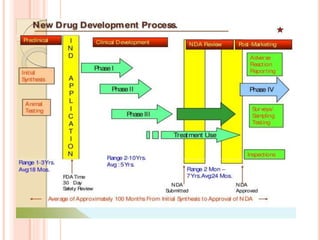
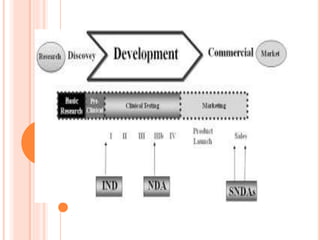
An Investigational New Drug (IND) application is required for clinical trials of any new drug or biological product. The IND application contains information on preclinical studies, manufacturing, protocols, investigator information and more. It allows the FDA to monitor clinical trials and place a hold if needed. Clinical trials are divided into Phases I-III to test safety, efficacy and dosing. The IND holder, sponsor and investigators all have responsibilities to protect subjects and ensure proper conduct of the clinical trials. Amendments are made to the IND as more information becomes available.






















![establishing_pv_centers_in_industry_AND_NATIONAL_PROGRAMME[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/establishingpvcentersinindustryandnationalprogramme1-230725101256-d16cc241-thumbnail.jpg?width=640&height=640&fit=bounds)

















































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)








