Download to read offline
































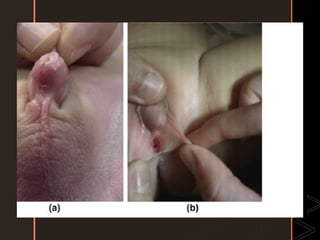








![• Is a disorder characterized by the presence of a higher level of
methemoglobin in the blood.
• Methemoglobin is an oxidized form of hemoglobin, {the iron
within hemoglobin is oxidized from the ferrous (Fe2+) state to
the ferric (Fe3+) state}, that has no affinity for oxygen,
resulting in no oxygen delivery to the tissue, so hypoxia can
occur.
• Clinically, this condition causes cyanosis.
• The major cause of inborn is glucose-6-phosphate
dehydrogenase [G6PD] deficiency and cytochrome b5 oxidase
deficiency) or severe acidosis, which impairs the function of
cytochrome b5 oxidase.
• This is particularly evident in young infants with diarrhea, in
whom excessive stool bicarbonate loss leads to metabolic
acidosis.
47](https://image.slidesharecdn.com/inbornerrorsofmetabolism-120429124218-phpapp0141-200506181549/85/Inbornerrorsofmetabolism-120429124218-phpapp01-4-1-47-320.jpg)

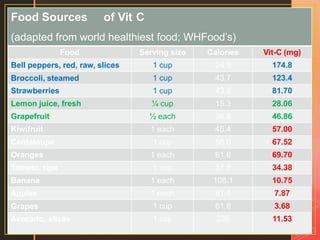

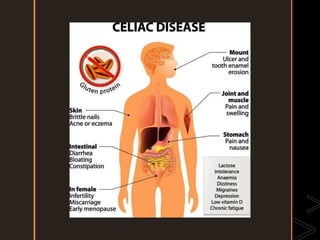


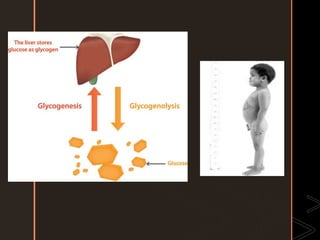
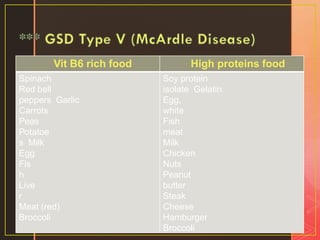
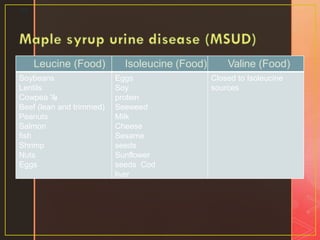
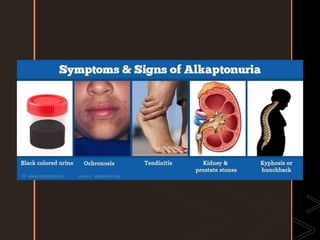
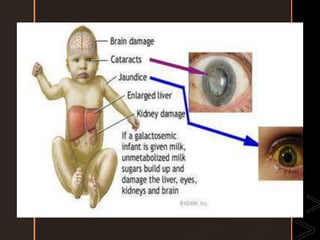
Inborn errors of metabolism are genetic disorders involving defects in metabolic pathways. Common types include disorders of carbohydrate metabolism like glycogen storage disease, amino acid metabolism like phenylketonuria, and organic acid metabolism. Symptoms result from substrate accumulation or product deficiency. Treatment focuses on dietary management to control metabolite levels and prevent complications like hypoglycemia, organ damage, and neurological decline. Strict dietary therapy and monitoring are needed lifelong for many of these inherited metabolic diseases.






















































![Human genome project [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/humangenomeprojectautosaved-210929062307-thumbnail.jpg?width=640&height=640&fit=bounds)
































