This document describes two case reports of patients with atypical benign childhood epilepsy with centrotemporal spikes (BECTS) who were also diagnosed with homocystinuria.
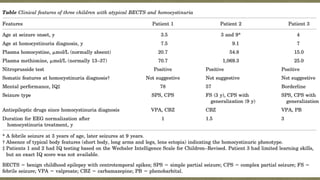
Patient 1 experienced seizures characterized by right arm jerking and speech arrest from age 3.5. Testing at age 6 showed BECTS on EEG but homocystinuria was not diagnosed until 18 months later. Treatment for homocystinuria resolved her seizures.
Patient 2 had a single febrile seizure at age 3 and two afebrile seizures at age 9 with EEG also showing BECTS. He was later found to have homocystinuria biochemically. Both patients showed improvement of neurological symptoms with









































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)

















