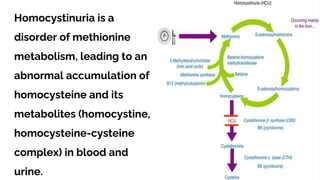
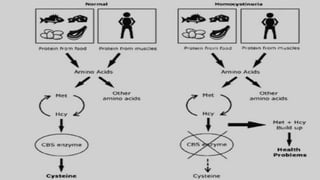
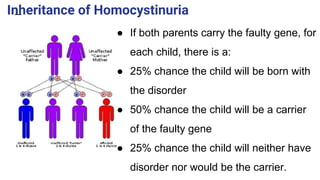
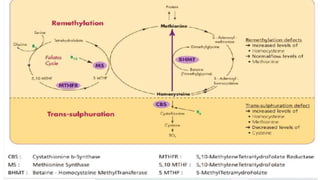
Homocystinuria is a hereditary metabolic disorder characterized by the accumulation of homocysteine due to defects in the enzyme cystathionine beta synthase, leading to various health complications affecting the eyes, skeletal system, central nervous system, and vascular system. It is inherited in an autosomal recessive manner with a prevalence of 1 in 200,000 to 350,000 live births, and symptoms can vary widely, including developmental delays, visual problems, physical deformities, mental retardation, and higher risks of cardiovascular issues. Diagnosis involves testing for elevated levels of methionine and homocystine, and although there is no cure, management includes lifelong vitamin supplementation and a low-methionine diet.