Downloaded 278 times





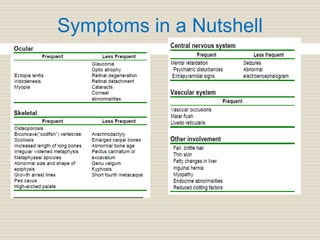


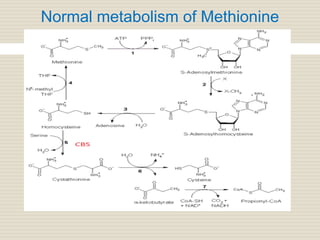

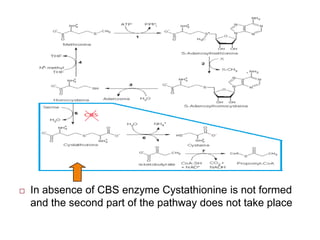






Homocystinuria is an inherited disorder caused by the inability to metabolize methionine due to a deficiency of the enzyme cystathionine beta synthase. This leads to an accumulation of homocysteine in the blood and urine. Symptoms affect the eyes, skeleton, central nervous system, and vascular system and include lens dislocation, bone abnormalities, intellectual disability, and blood clots. Treatment involves a low-methionine diet, vitamin B6, betaine, and monitoring methionine levels. Genetic counseling and newborn screening can help detect the condition early to prevent complications.























































































