Downloaded 110 times





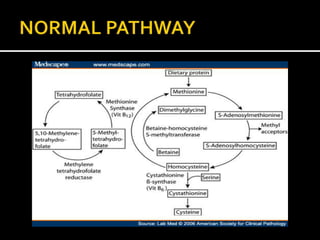

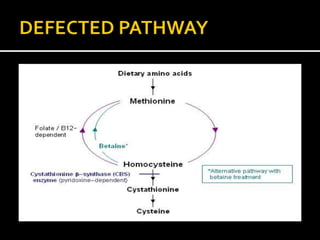




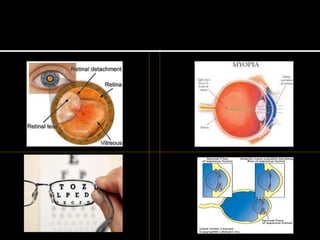

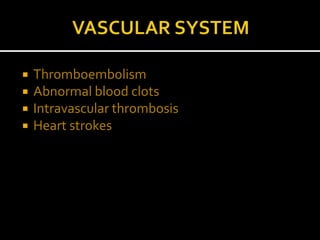
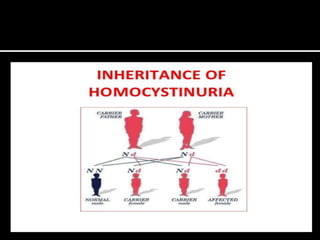











Homocystinuria is an inherited disorder caused by the body's inability to break down the amino acid homocysteine. This leads to an accumulation of homocysteine in the blood and urine. Symptoms involve the eyes, bones, brain and blood vessels, and can include dislocated lenses, bone abnormalities, intellectual disability, and blood clots leading to heart attacks or strokes. Treatment aims to reduce homocysteine levels through a low-methionine diet, vitamin supplements like B6, B12, and betaine.























































































