Downloaded 22 times
















![PHYSIOLOGICAL VARIETIES OF HAEMOGLOBIN
• Adult haemoglobin or haemoglobin A [HbA (α2β2)]
Two types:
(i) Haemoglobin A [HbA (α2β2)]
• The main form of normal adult Hb
• Its globin part consists of two α and two β polypeptide chains
• It is a spheroidal molecule with a molecular weight of 68,000
(ii) Haemoglobin A2 [HbA2 (α2δ2)]
• Minor component (about 2.5% of the total Hb) in normal adults
• Globin part consists of two α and two δ polypeptide chains.
• δ chains slightly different amino acid composition (out of 146, 10 amino acids are
different) as compared to β chains.](https://image.slidesharecdn.com/hemoglobin2-200617051941/85/Hemoglobin-2-18-320.jpg)
![PHYSIOLOGICAL VARIETIES OF HAEMOGLOBIN
Fetal haemoglobinor haemoglobin F [HbF (α2γ2)]
• Hb present in the fetal RBCs
• disappears 2–3 months after birth
Structure of HbF:
• Globin part consistsof two α and two γ polypeptide chains (in place of β chains).
• γ chains also have 146 amino acids but its 37 amino acids are different than that of β
chains
Special features of HbF :
• Affinity for oxygen in case of HbF is more than that of HbA
• Resistanceto action of alkalies is more in HbF than HbA
• Life span of HbF is much less (1–2 week) as compared to that of HbA (120 days)](https://image.slidesharecdn.com/hemoglobin2-200617051941/85/Hemoglobin-2-19-320.jpg)


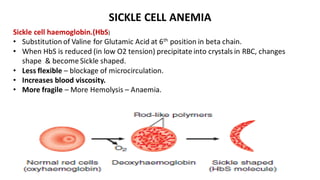
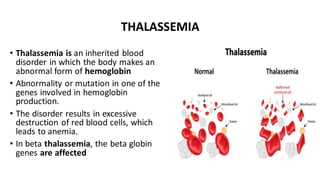












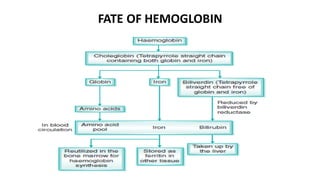




































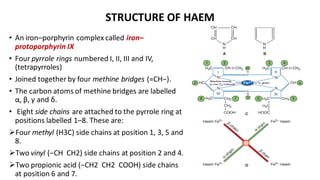
Hemoglobin is the iron-containing protein in red blood cells that transports oxygen throughout the body. It is composed of four polypeptide chains and an iron-containing heme group. Normal hemoglobin levels vary based on age and sex. Hemoglobin transports oxygen from the lungs to tissues and carbon dioxide from tissues back to the lungs. Abnormal hemoglobin variants can lead to conditions like sickle cell anemia and thalassemia. Red blood cells have an average lifespan of 120 days before being broken down, mainly in the spleen.



































































