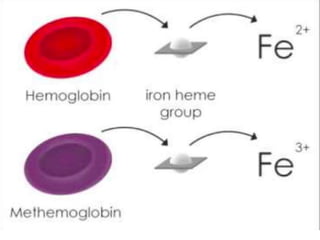
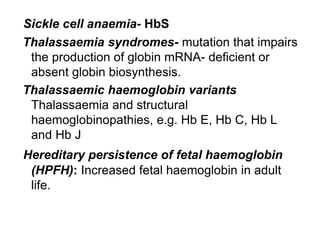
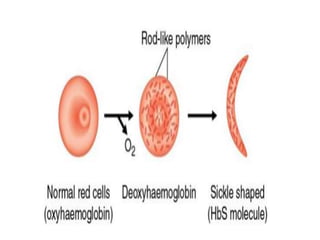
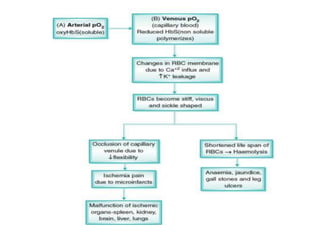
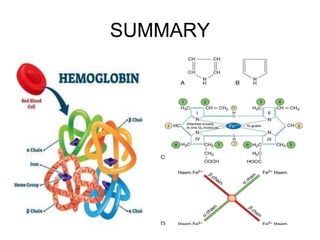
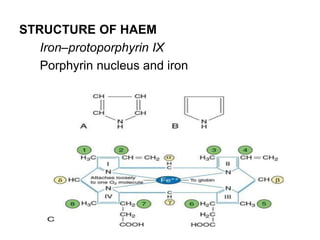
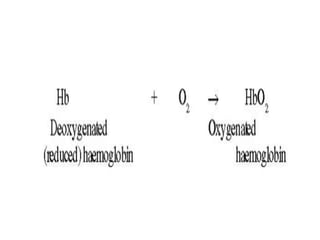
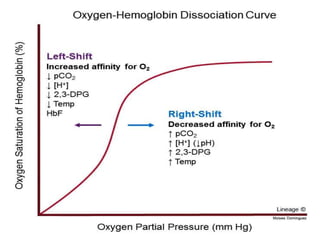
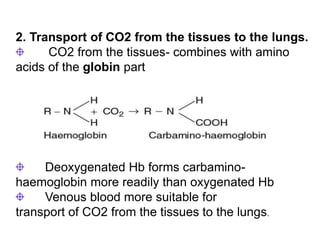
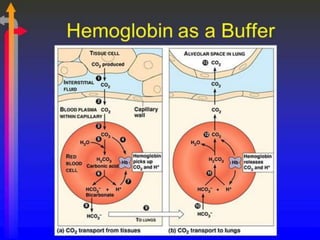
This document provides information on hemoglobin structure and function. It discusses the history of hemoglobin discovery and describes the globin and heme components. Hemoglobin transports oxygen from the lungs to tissues and carbon dioxide from tissues to the lungs. Varieties include adult, fetal, and other variants like hemoglobin S, C, and E. Derivatives such as oxyhemoglobin, carbaminohemoglobin, and methemoglobin are also reviewed. Hemoglobinopathies include structural variants and thalassemias. Sickle cell anemia results from substitution of one amino acid, causing red blood cells to sickle in low oxygen conditions. Thalassemias are globin chain synthesis defects.























![PHYSIOLOGICAL VARIETIES OF
HAEMOGLOBIN
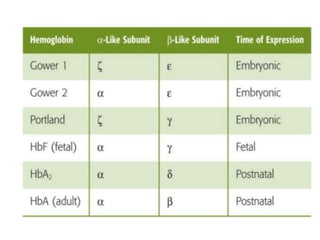
ADULT HAEMOGLOBIN:
It is of two types.
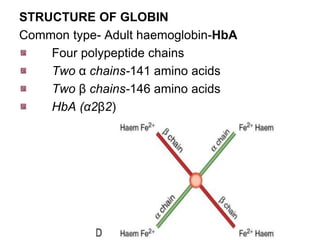
Haemoglobin A [HbA (α2β2)]- normal adult
haemoglobin
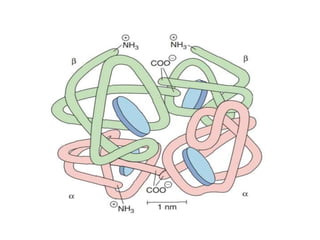
Globin part consists- two alpha and two beta
polypeptide chains
It is a spheroidal molecule
molecular weight- 68,000.](https://image.slidesharecdn.com/hbseminarnew-210221102228/85/HAEMOGLOBIN-STRUCTURE-FUNCTION-33-320.jpg)
![ii. Haemoglobin A2 [HbA2 (α2δ2)]
Minor component- 2.5% of the total Hb in
normal adults.
Globin part-two alpha and two delta
polypeptide chains.
Delta chains- out of 146, 10 amino acids are
different compared with β chains](https://image.slidesharecdn.com/hbseminarnew-210221102228/85/HAEMOGLOBIN-STRUCTURE-FUNCTION-34-320.jpg)
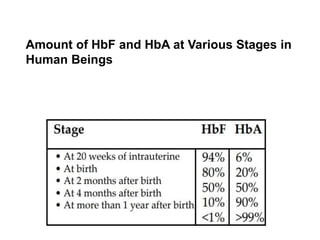
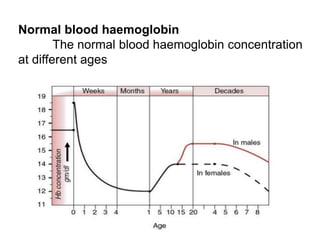
![FETAL HAEMOGLOBIN:
Haemoglobin F [HbF α2γ2] - fetal RBCs
It gradually disappears 2–3 months after birth](https://image.slidesharecdn.com/hbseminarnew-210221102228/85/HAEMOGLOBIN-STRUCTURE-FUNCTION-36-320.jpg)