This document summarizes key points from a workshop on machine learning for materials science held on August 1, 2019. It discusses how graphs are a natural representation for materials as they can capture local atomic environments and periodicity in crystals. A graph network framework called MEGNet is presented that achieves state-of-the-art performance for molecular and crystal property prediction. MEGNet models outperform previous methods on standard benchmarks and allow for transfer learning across properties. The workshop also covered practical considerations for training deep learning models and applications beyond bulk crystals.

![High-throughput computation is not enough:A
statistical history of the Materials Project
Aug 1 2019
Reasonable ML
Deep learning
(AA’)0.5(BB’)0.5O3 perovskite
2 x 2 x 2 supercell,
10 A and 10 B species
= (10C2 x 8C4)2 ≈107
NIST Workshop
ratio of (634 + 34)/485 ≈ 1.38 (Supplementary Table S-II) with b5%
difference in the experimental and theoretical values. This again
agree well with those calculated from the rule of mixture (Supplemen-
tary Table-III). The experimental XRD patterns also agree well with
Fig. 2. Atomic-resolution STEM ABF and HAADF images of a representative high-entropy perovskite oxide, Sr(Zr0.2Sn0.2Ti0.2Hf0.2Mn0.2)O3. (a, c) ABF and (b, d) HAADF images at (a, b) low
and (c, d) high magnifications showing nanoscale compositional homogeneity and atomic structure. The [001] zone axis and two perpendicular atomic planes (110) and (110) are marked.
Insets are averaged STEM images.
Jiang et al. A New Class of
High-Entropy Perovskite
Oxides. Scripta Materialia
2018, 142, 116–120.
Materials design is
combinatorial](https://image.slidesharecdn.com/shyuepingong-nistworkshop-190826170819/85/Graphs-Environments-and-Machine-Learning-for-Materials-Science-2-320.jpg)

















![Machine learning the potential energy surface
Aug 1 2019 NIST Workshop
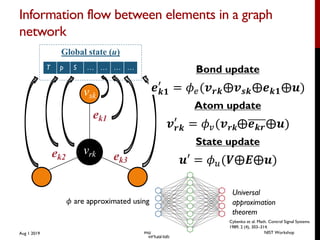
Local environment descriptors ML approach
A separate neural network is used for each atom. The neural network is defined by
the number of hidden layers and the nodes in each layer, while the descriptor space is
given by the following symmetry functions:
Gatom,rad
i =
NatomX
j6=i
e ⌘(Rij Rs)2
· fc(Rij),
Gatom,ang
i = 21 ⇣
NatomX
j,k6=i
(1 + cos ✓ijk)⇣
· e ⌘0(R2
ij+R2
ik+R2
jk)
· fc(Rij) · fc(Rik) · fc(Rjk),
where Rij is the distance between atom i and neighbor atom j, ⌘ is the width of the
Gaussian and Rs is the position shift over all neighboring atoms within the cuto↵
radius Rc, ⌘0
is the width of the Gaussian basis and ⇣ controls the angular resolution.
fc(Rij) is a cuto↵ function, defined as follows:
fc(Rij) =
8
>><
>>:
0.5 · [cos (
⇡Rij
Rc
) + 1], for Rij Rc
0.0, for Rij > Rc.
These hyperparameters were optimized to minimize the mean absolute errors of en-
ergies and forces for each chemistry. The NNP model has shown great performance
for Si,11
TiO2,40
water41
and solid-liquid interfaces,42
metal-organic frameworks,43
and
has been extended to incorporate long-range electrostatics for ionic systems such as
4
Atom-centered symmetry
functions (ACSF)
Moment tensors
Smooth overlap of atomic
positions (SOAP)
SO4 bispectrum
Polynomial / Linear
regression
Kernel regression
Neural networks
ZnO44
and Li3PO4.45
2. Gaussian Approximation Potential (GAP). The GAP calculates the similar-
ity between atomic configurations based on a smooth-overlap of atomic positions
(SOAP)10,46
kernel, which is then used in a Gaussian process model. In SOAP, the
Gaussian-smeared atomic neighbor densities ⇢i(R) are expanded in spherical harmonics
as follows:
⇢i(R) =
X
j
fc(Rij) · exp(
|R Rij|2
2 2
atom
) =
X
nlm
cnlm gn(R)Ylm( ˆR),
The spherical power spectrum vector, which is in turn the square of expansion coe -
cients,
pn1n2l(Ri) =
lX
m= l
c⇤
n1lmcn2lm,
can be used to construct the SOAP kernel while raised to a positive integer power ⇣
(which is 4 in present case) to accentuate the sensitivity of the kernel,10
K(R, R0
) =
X
n1n2l
(pn1n2l(R)pn1n2l(R0
))⇣
,
In the above equations, atom is a smoothness controlling the Gaussian smearing, and
nmax and lmax determine the maximum powers for radial components and angular com-
ponents in spherical harmonics expansion, respectively.10
These hyperparameters, as
well as the number of reference atomic configurations used in Gaussian process, are
Behler-Parinello Neural
Network Potential (NNP)1
Moment Tensor Potential
(MTP)2
Gaussian Approximation
Potential (GAP)3
Spectral Neighbor Analysis
Potential (SNAP)4
ACSF/MT encodes distances and angles.
SOAP/bispectrum encodes neighbor density.
Interatomic Potential
1 Behler et al. PRL. 98.14 (2007): 146401.
2 Shapeev MultiScale Modeling and Simulation 14, (2016).
3 Bart ́ok et al. PRL. 104.13 (2010): 136403.
4 Thompson et al. J. Chem. Phys. 285, 316330 (2015)](https://image.slidesharecdn.com/shyuepingong-nistworkshop-190826170819/85/Graphs-Environments-and-Machine-Learning-for-Materials-Science-21-320.jpg)


![ML-IAP:Accuracy vs Cost
Aug 1 2019 NIST Workshop
Testerror(meV/atom)
Computational cost s/(MD step atom)
a
b
Jmax = 3
Jmax = 3
2000 kernels20 polynomial powers
hidden layers [16, 16]
GAP reaches
best accuracy,
but is the most
expensive by
O(102-103)
MTP, NNP,
qSNAP all lie
quite close to
Pareto frontier.
Mo dataset
Zuo et al. A Performance and Cost Assessment of Machine Learning Interatomic Potentials. arXiv:1906.08888 2019.](https://image.slidesharecdn.com/shyuepingong-nistworkshop-190826170819/85/Graphs-Environments-and-Machine-Learning-for-Materials-Science-24-320.jpg)


![Applications:Ni-Mo phase diagram and
mechanical behavior
Aug 1 2019 NIST Workshop
Solid-liquid equilibrium Hall-Petch strengthening
[1] Hu et al. Nature, 2017, 355, 1292](https://image.slidesharecdn.com/shyuepingong-nistworkshop-190826170819/85/Graphs-Environments-and-Machine-Learning-for-Materials-Science-27-320.jpg)










































![241223_JW_labseminar[Neural Message Passing for Quantum Chemistry].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/241223jwlabseminarmpnn-241223090919-ea3d6132-thumbnail.jpg?width=640&height=640&fit=bounds)