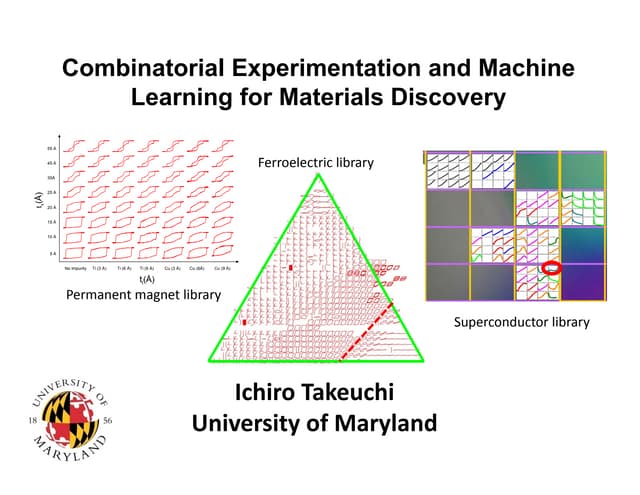
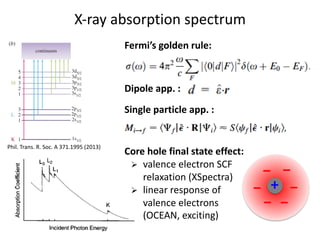
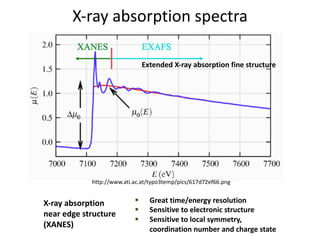
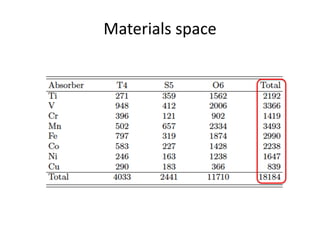
The document discusses using theory, computation, and machine learning to interpret experimental X-ray absorption spectroscopy data and determine local atomic structures. It presents examples of using density functional theory calculations of X-ray absorption near edge structure (XANES) spectra to benchmark predictions against experiments and develop machine learning models for structure classification. The models are able to classify local structures like tetrahedral, square pyramidal, and octahedral coordination with over 85% accuracy across different materials systems. This approach provides a way to solve the inverse problem of determining structures from spectroscopy measurements in real time.








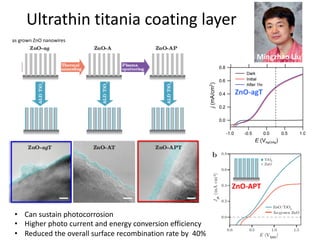
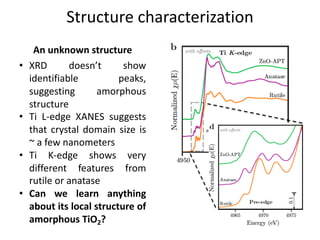
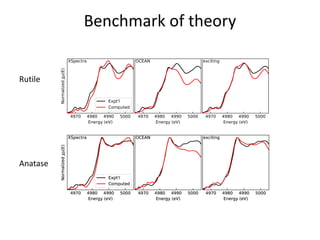
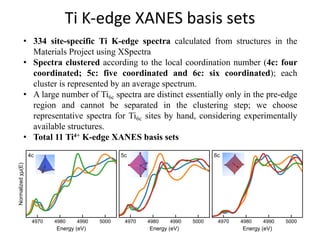





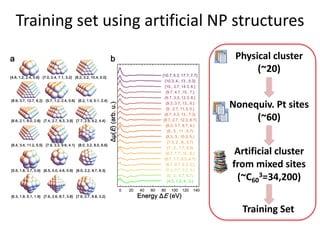

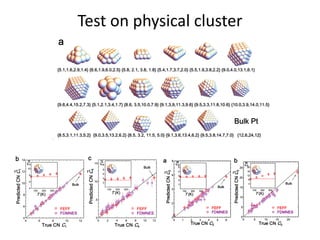
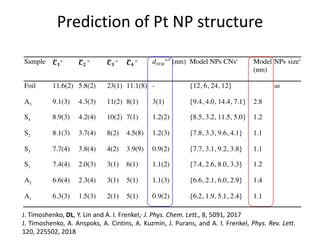

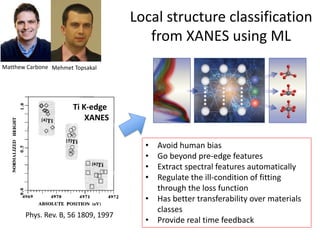



























![nanotechnology-ppt[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/nanotechnology-ppt1-221201180129-04da0508-thumbnail.jpg?width=640&height=640&fit=bounds)