Downloaded 197 times



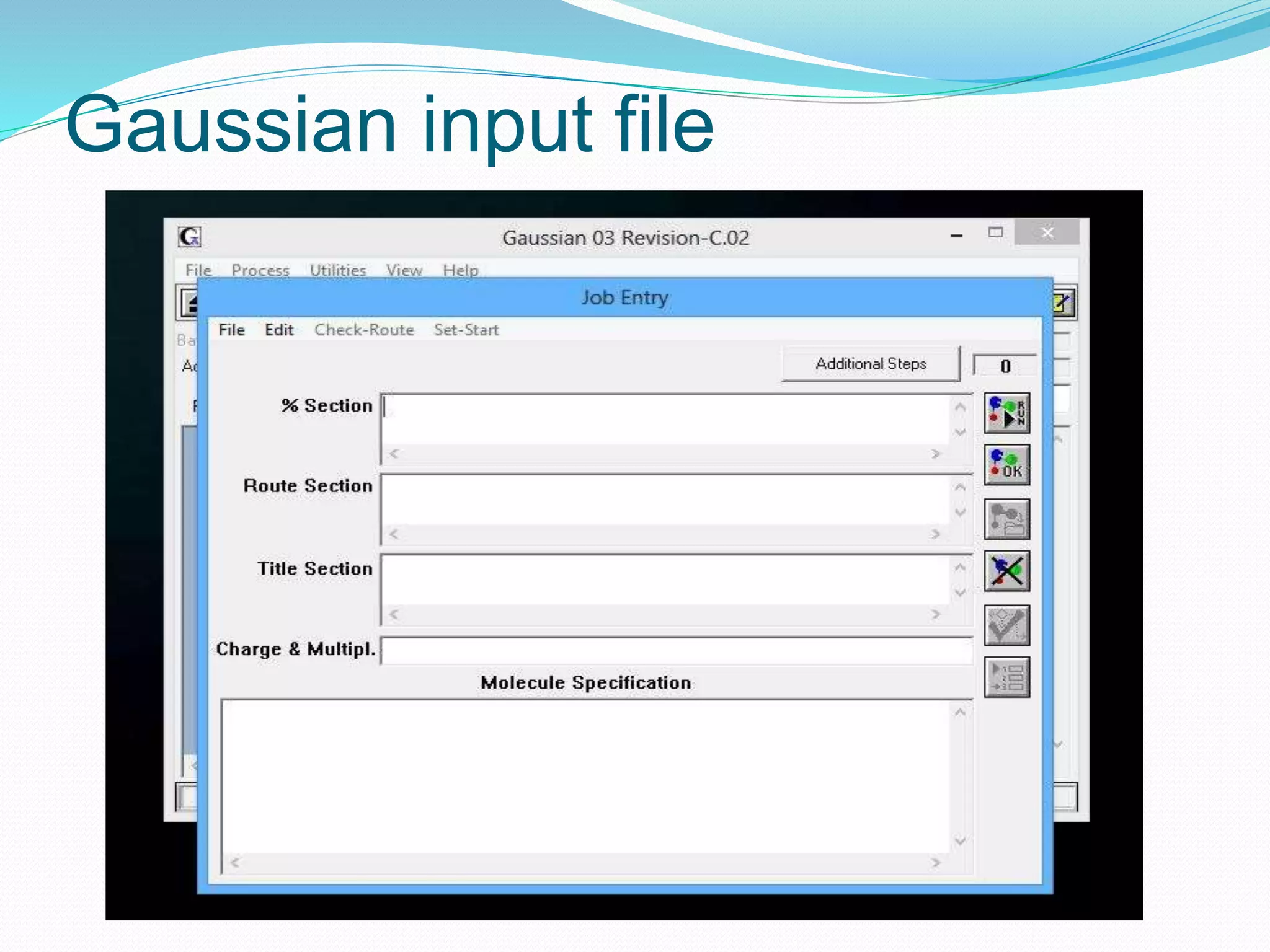














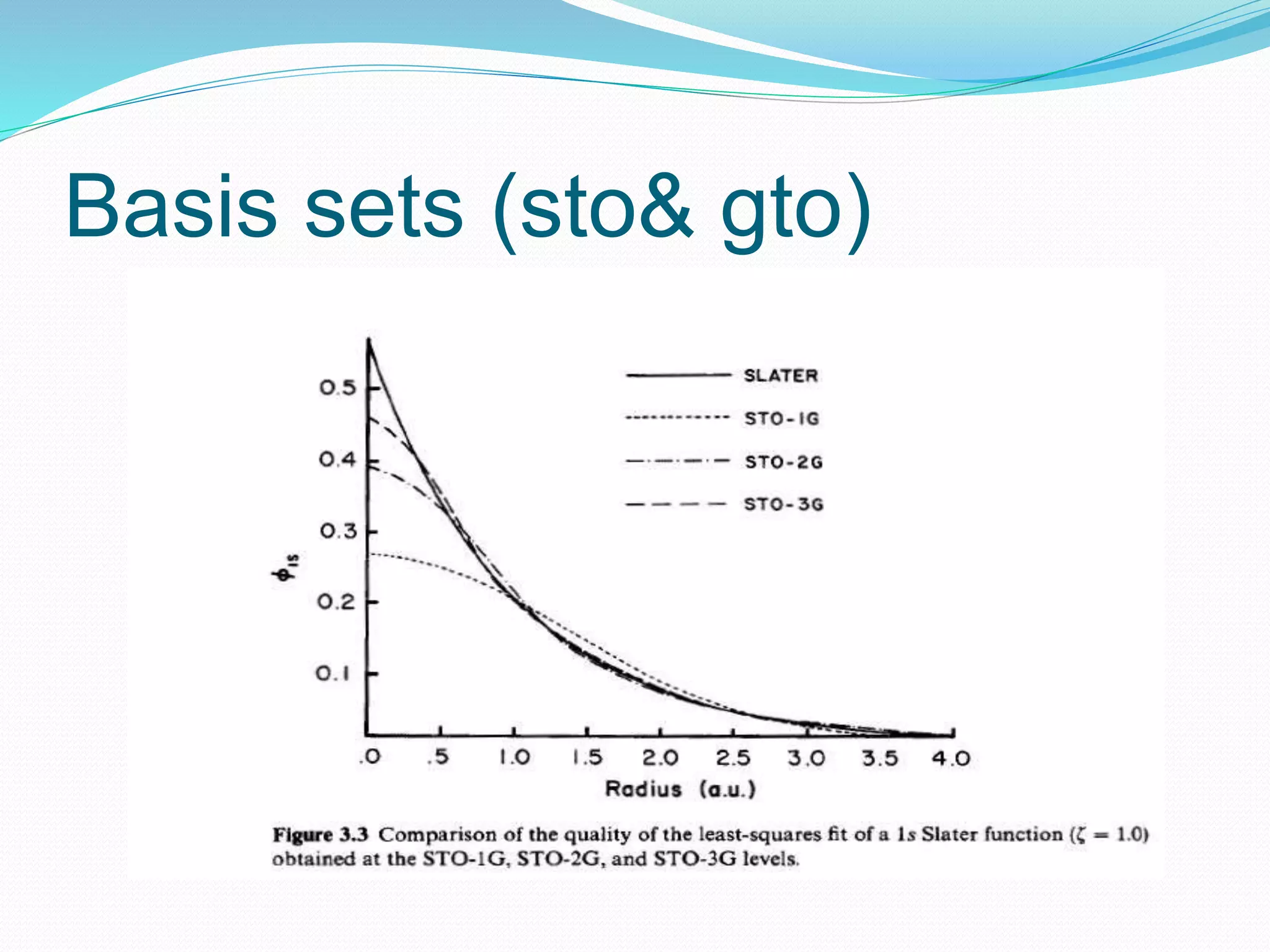









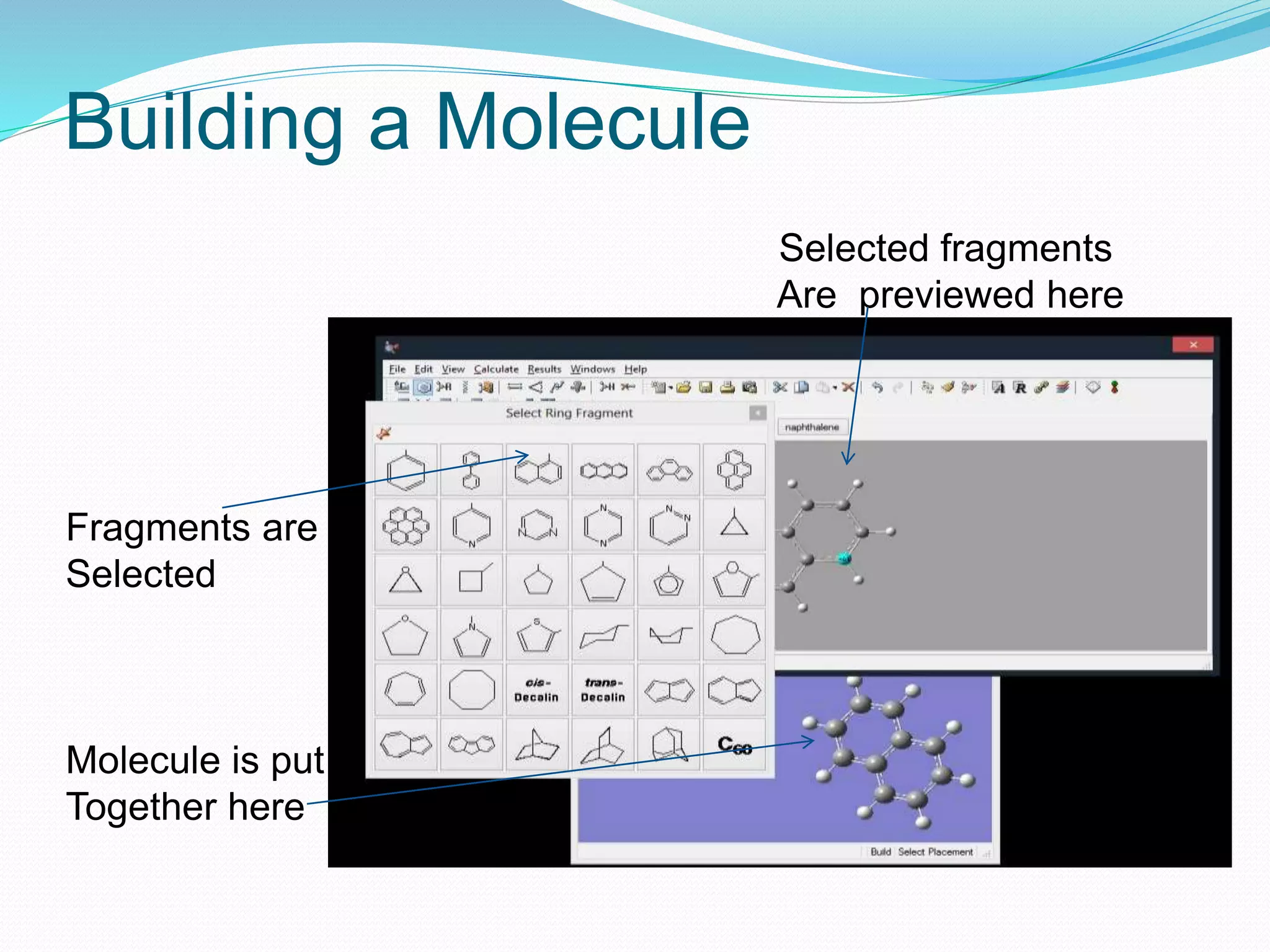

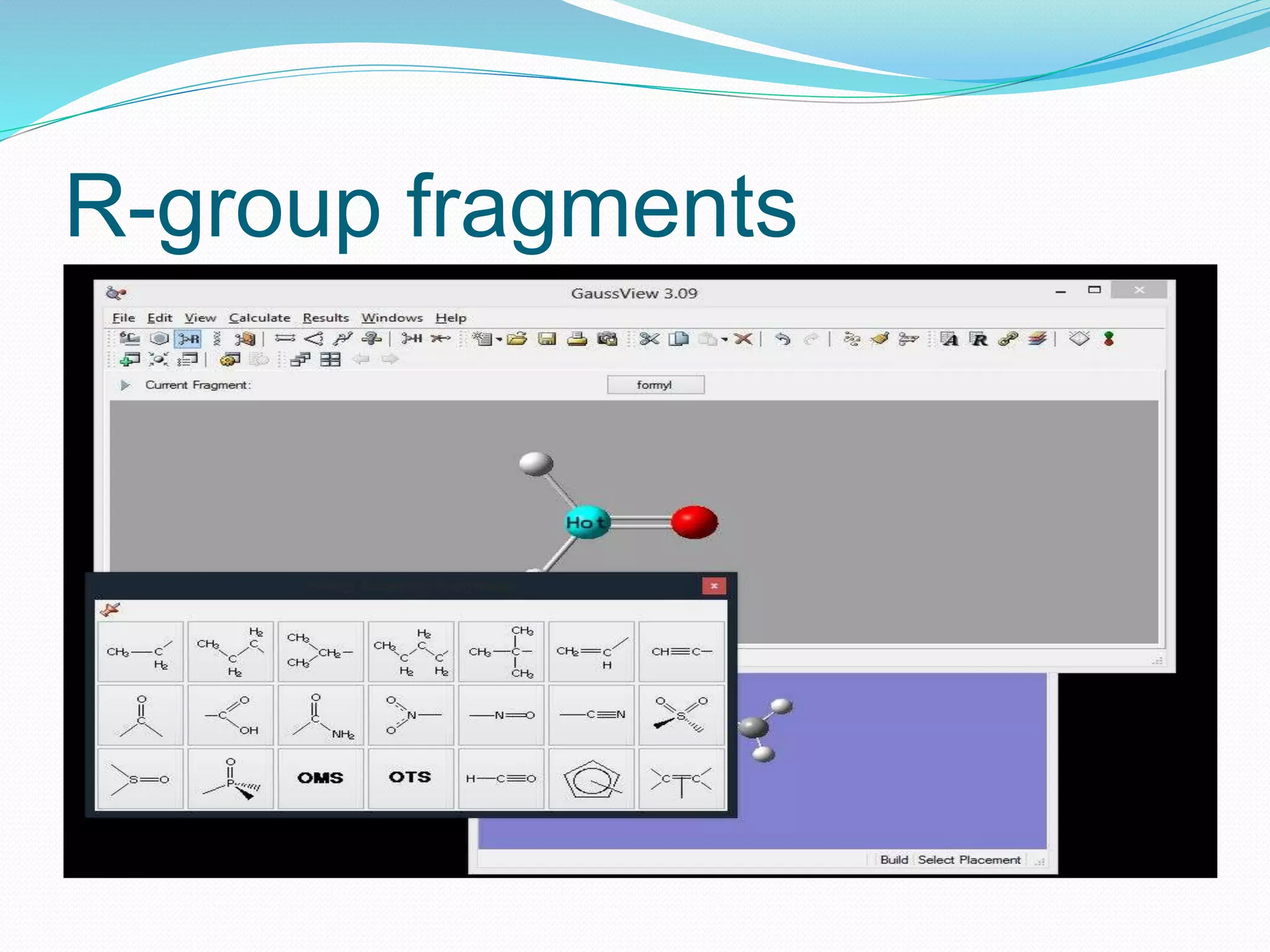










Gaussian is capable of performing several quantum chemical calculations including molecular energies, geometry optimization, vibrational frequencies, NMR properties, potential energy surfaces, and reaction pathways. It takes a Gaussian input file specifying the calculation type, theory, basis set, coordinates, etc. Common calculation types include single point energy, geometry optimization, and vibrational frequency. The output file provides optimized geometry, frequencies, energies, and other molecular properties.



































































