Download as PDF, PPTX



![The Problem
Motivation: Top 20 best-selling drugs
in America had sales of ~ $65bn in
2005[1]
New drug development costs are in
excess of $800M[2]
Roughly 10K structures are made and
tested for every new drug reaching the
market[3]
[1] The Best-Selling drugs in America, IMS health, 2006
[2] The Tufts Center for the study of drug development
[3] Boston Consulting Group, 2005](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-4-320.jpg)



![Gas phase water: An example
A DFT calculation
takes ~9s
An “Exact”
calculation[4] took
150h, 250Gb of
memory, and 800Gb
of disk
[4] G. K.-L. Chan and M. Head-Gordon, J. Chem. Phys. 118, 8551 2003](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-9-320.jpg)

![The Hospital that ate my Wife. . .
Information theoretic properties of a model system:
Sr = " $ # (r) ln[ #(r)]dr
S p = " $ % (p) ln[% (p)]dp
ST = Sr + S p
Doesn’t Sr look a little familiar?
!](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-11-320.jpg)
![A novel descriptor?
Continuous form of a measure used in molecular
similarity:
S = "# pi ln[ pi ]
i
Could we use Sr as a measure of similarity?
Moreover, could Sr be a 3D QM-based
!
structural descriptor?
– Literature search has shown that this has not been
considered before (I think)[5]
[5] M. Karelson, “Quantum-chemical descriptors in QSAR”, in Computational Medicinal
Chemistry for Drug Discovery, P. Bultnick et al, Eds., (New York, Dekker, 2003), pp 641-667](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-12-320.jpg)
![A novel descriptor?
We want to make this useful
– But we still have the problem of finding ρ in a
timely fashion
Why don’t we approximate ρ?
– We construct a pro-molecular density from a sum
of fitted s-Gaussians[6]
"(r) # " Mol (r) = % "$ (r) = % % c$i exp(&'$i (r & R$ ) 2 )
$ $ i
Turns out that this isn’t as bad as you might
think[7]
! P. Constans and R. Carbó, J. Chem. Inf. Sci. 35, 1046 1995
[6]
[7] J. I. Rodriguez, D. C. Thompson, and P. W. Ayers Unpublished data](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-13-320.jpg)
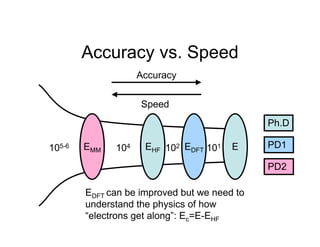
![Homebrew quantum mechanics
All of this has been done on my iMac at home
Molecular integrations performed using the
Becke/Lebedev grids in PyQuante[8]
Co-opted graduate students into doing
MathCad checks for me. . .
[8] Python Quantum Chemistry - http://pyquante.sourceforge.net/](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-14-320.jpg)
![Homebrew quantum mechanics
Molecule Sr
H2O -7.42
H2S 3.94
Benzene -27.09
Cyclohexane (chair) -35.94
Perhaps Sr isn’t that discriminatory?
Plan B - Sr (r) = " #(r)ln[ # (r)]](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-16-320.jpg)




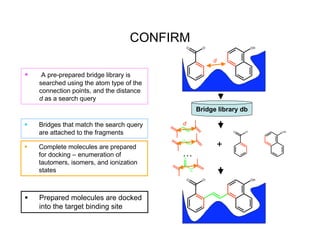
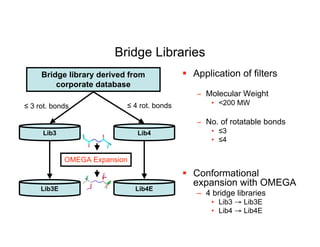
![CONFIRM: Novelty
Bridges come from molecules within the Wyeth
CORP database:
– Bridges obtained “…from a given ring scaffold by removing
all of the atoms, except acyclic linker atoms, between pairs
of ring systems, and the anchor atoms on the ring system.”
[9]
Similar to CAVEAT[10], however:
– We do not use orientation of bonds, but location of atoms
(vector vs. scalar)
– CAVEAT searches 3D databases looking for suitable
molecular frameworks to satisfy the vector pairs
• We already have well defined positions of small molecule
binders
[9] R. Nilakantan et al., J. Chem. Inf. Mod. 46(3), 1069-1077 2006
[10] G. Lauri, and P. A. Bartlett, J. Comp.-Aided. Mol. Design 8(1), 51-66 1994](https://image.slidesharecdn.com/thompsondc-bi-110831085017-phpapp02/85/Computational-Chemistry-From-Theory-to-Practice-27-320.jpg)

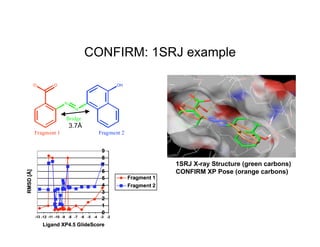
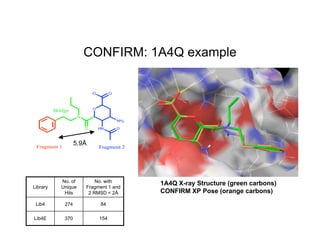
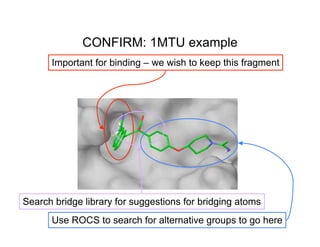



The document discusses computational chemistry's applications in drug design, particularly focusing on the introduction of a novel 3D quantum mechanics-based structural descriptor and its potential utility. It covers challenges in new drug development, such as high costs and extensive structures needed for market approval, and presents methods for fragment-based de novo design to explore the vast chemical space efficiently. The document emphasizes the importance of understanding electron density for accurate calculations and the development of algorithms for linking and growing molecular fragments.























































































