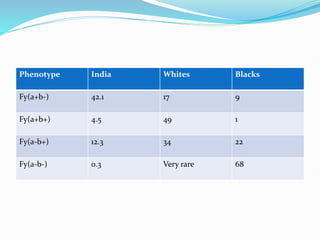
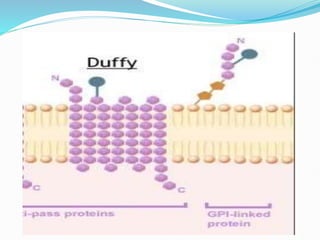
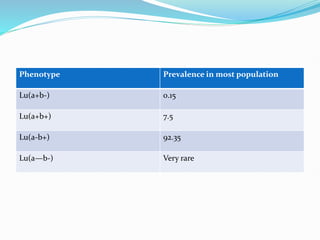
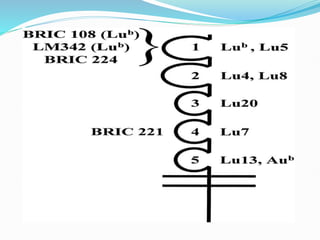
The Duffy blood group system is an important blood group system that impacts susceptibility to malaria. It was discovered in the 1950s through studies of patients who developed antibodies. The Duffy antigens Fya and Fyb are carried on the Duffy glycoprotein, which acts as a receptor for malaria parasites and chemokines. People with the Fy(a-b-) phenotype, common in Africans, lack the Duffy glycoprotein and are resistant to malaria. The document provides details on the genetics and biochemistry of the Duffy glycoprotein, as well as the various Duffy antigens and antibodies that have been discovered.























































































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)




