[DL輪読会]“Highly accurate protein structure prediction with AlphaFold”
1.
1
DEEP LEARNING JP
[DLPapers]
http://deeplearning.jp/
“Highly accurate protein structure prediction with AlphaFold”
Kensuke Wakasugi, Panasonic Corporation.
2.
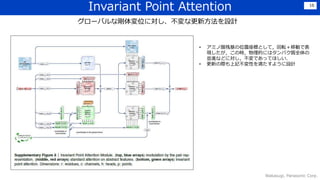
紹介論文
タイトル:Highly accurate proteinstructure prediction with AlphaFold
著者:Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., ...
& Hassabis, D. 合計33人.
所属:DeepMind、ソウル大学校
その他: Nature掲載(2021/07/15公開),引用82件(2021/09/06時点)
https://www.nature.com/articles/s41586-021-03819-2
選書理由
AlphaFoldの原型は昨年時点で公開されていたが,その進化版としてAlphaFold2が話題に
なっていたため
書誌情報
Wakasugi, Panasonic Corp.
2
※本資料の図表は,特に記載がない限り紹介論文より引用したものです
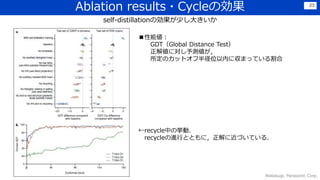
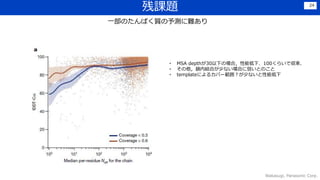
Discussion
Wakasugi, Panasonic Corp.
25
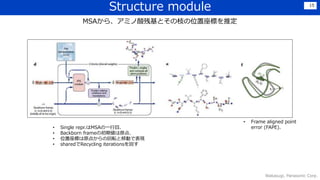
暗黙的な物理制約を自然に獲得できている
•バイオインフォと物理的観点から手法を構築することで,最小限のマニュアル特徴量でも、うまく構造の性質をつかむことができた
→水素の結合長などは,陽に含んでいないが,うまく予測できている
• 欠落した物理的背景もうまく推論できており,intertwined homomers(アミノ酸配列が同一単位の繰り返しで構成され、全体が絡み
合ったもの?)もうまく予測できている(下図)
• たんぱく質の必須解析ツールになることを期待.※github公開.有志によるブラウザ経由のツールもある
同じグループから,解析よりの論文もすでに発表されている
Highly accurate protein structure prediction for the human proteome
https://www.nature.com/articles/s41586-021-03828-1
![1
DEEP LEARNING JP
[DL Papers]
http://deeplearning.jp/
“Highly accurate protein structure prediction with AlphaFold”
Kensuke Wakasugi, Panasonic Corporation.](https://image.slidesharecdn.com/210910dlver11-210910034033/85/DL-Highly-accurate-protein-structure-prediction-with-AlphaFold-1-320.jpg)
![1
DEEP LEARNING JP
[DL Papers]
http://deeplearning.jp/
“Highly accurate protein structure prediction with AlphaFold”
Kensuke Wakasugi, Panasonic Corporation.](https://image.slidesharecdn.com/210910dlver11-210910034033/75/DL-Highly-accurate-protein-structure-prediction-with-AlphaFold-1-2048.jpg)


![CASPとは
■CASP:The Critical Assessment of protein Structure Prediction[1]
→タンパク質構造予測のコンペティション.1994年から2年おきに開催.CASP14は2020年開催.
Wakasugi, Panasonic Corp.
4
[1]CASP:https://predictioncenter.org/index.cgi 訪問日2021/09/06
[2]フォールディング 出典: フリー百科事典『ウィキペディア(Wikipedia)』訪問日2021/09/06
カテゴリ 内容
テンプレートベース 既知構造を用いて,構造予測
テンプレートフリー いちから構造予測
接触予測 部分構造の接触を予測
構造生物学への応用 未知構造への応用
精密化 後処理による構造の精緻化?
実験とのハイブリッド 低解像度の実験計測との組み合わせ
メイン
タンパク質のフォールディング[2]
ヒトの場合、
20種のアミノ
酸の配列が入力
入力 出力
CASPカテゴリ[1]](https://image.slidesharecdn.com/210910dlver11-210910034033/85/DL-Highly-accurate-protein-structure-prediction-with-AlphaFold-4-320.jpg)
![CASP14@2020
Wakasugi, Panasonic Corp.
5
[1]CASP HP:https://predictioncenter.org/index.cgi 訪問日2021/09/06
[1]より引用
実験精度
全体の2/3
高精度
全体の90%
CASP14にてAlphaFold2が大幅に精度向上
約90%のタンパク質の構造を正確に予測
AlphaFold2が実験精度に到達](https://image.slidesharecdn.com/210910dlver11-210910034033/85/DL-Highly-accurate-protein-structure-prediction-with-AlphaFold-5-320.jpg)
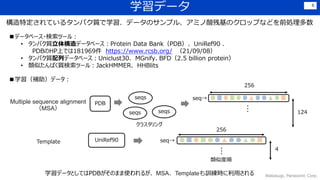
![学習データ 7
主に,sequence数 × residue数 × [アミノ酸onehot or 正解構造座標]
その他として,該当アミノ酸残基より
左にある欠失の数等が含まれる](https://image.slidesharecdn.com/210910dlver11-210910034033/85/DL-Highly-accurate-protein-structure-prediction-with-AlphaFold-7-320.jpg)
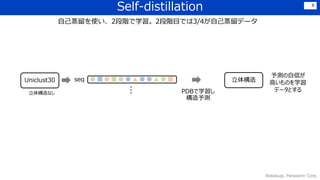
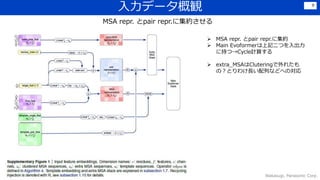
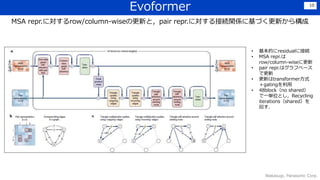





![感想
Wakasugi, Panasonic Corp.
26
• Transformerなどを利用しつつ,細部にわたって工夫を凝らしている印象.
Lossの組合せ、各種crop、データの前処理・選定、Recycle
• ゲノム解析 → タンパク質構造同定 → 機能分析 の流れが加速することが期待される
余談:
web記事[3]によると,AlphaFold2のファイル容量は2.2TBらしい
RoseTTAFoldという手法もワシントン大学から発表されているとのこと
[3]https://www.itmedia.co.jp/news/articles/2107/20/news136.html 訪問日2021/09/09](https://image.slidesharecdn.com/210910dlver11-210910034033/85/DL-Highly-accurate-protein-structure-prediction-with-AlphaFold-26-320.jpg)



![[DL輪読会]GQNと関連研究,世界モデルとの関係について](https://cdn.slidesharecdn.com/ss_thumbnails/20180817-180827085537-thumbnail.jpg?width=640&height=640&fit=bounds)


![[DL輪読会]Flow-based Deep Generative Models](https://cdn.slidesharecdn.com/ss_thumbnails/20190307-190328024744-thumbnail.jpg?width=640&height=640&fit=bounds)

![[DL輪読会]相互情報量最大化による表現学習](https://cdn.slidesharecdn.com/ss_thumbnails/20190913iwasawa-190913002312-thumbnail.jpg?width=640&height=640&fit=bounds)



![[DL輪読会]GLIDE: Guided Language to Image Diffusion for Generation and Editing](https://cdn.slidesharecdn.com/ss_thumbnails/glide2-220107030326-thumbnail.jpg?width=640&height=640&fit=bounds)



![[DL輪読会]Diffusion-based Voice Conversion with Fast Maximum Likelihood Samplin...](https://cdn.slidesharecdn.com/ss_thumbnails/20220318akuzawa-220322065615-thumbnail.jpg?width=640&height=640&fit=bounds)

![[DL輪読会]Learning Transferable Visual Models From Natural Language Supervision](https://cdn.slidesharecdn.com/ss_thumbnails/dlkobayashi0115-210115012308-thumbnail.jpg?width=640&height=640&fit=bounds)






![[DL輪読会]Neural Ordinary Differential Equations](https://cdn.slidesharecdn.com/ss_thumbnails/nnasode1-190111001755-thumbnail.jpg?width=640&height=640&fit=bounds)






















