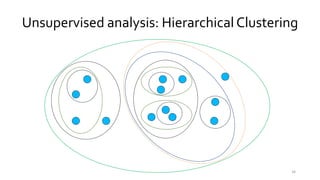
T-BioInfo is a platform for processing, analyzing, and integrating multi-omics data. It is used by multiple research groups to extract meaningful insights from large multi-omics datasets. The platform is expanding its educational capabilities to enable more people to extract meaningful, data-driven insights from omics datasets with biomedical applications. The document provides links to learn more about the platform's research and educational features.

























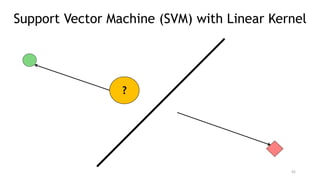




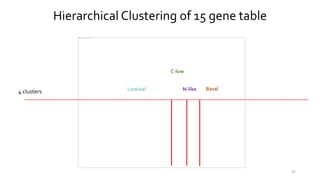



![52
http://www.oncotarget.com/index.php?journal=oncotarget&page=arti
cle&op=view&path[]=23869&path[]=75083
https://www.nature.com/articles/1208329](https://image.slidesharecdn.com/may-workshop1p-180514234815/85/May-15-workshop-52-320.jpg)