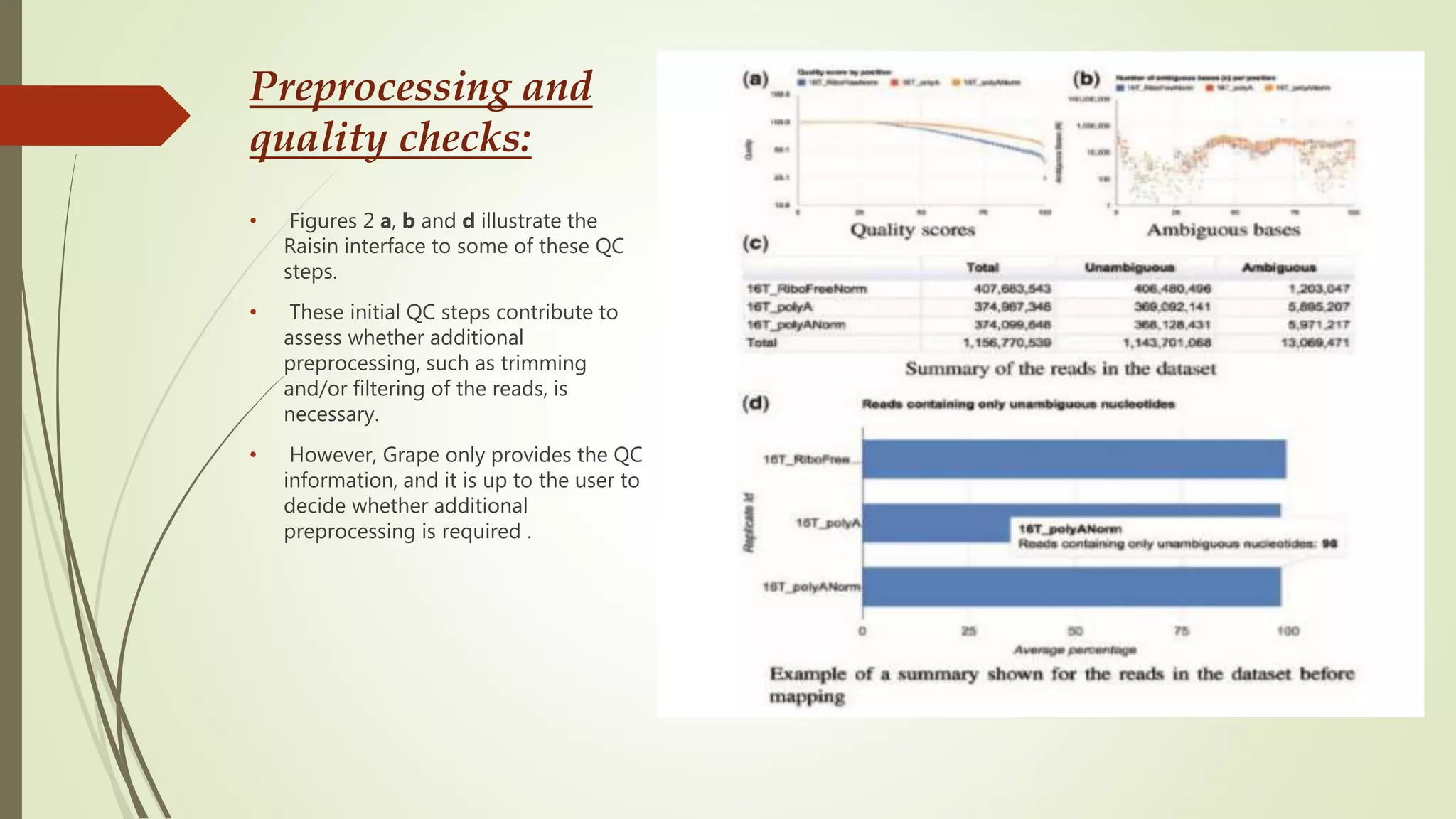
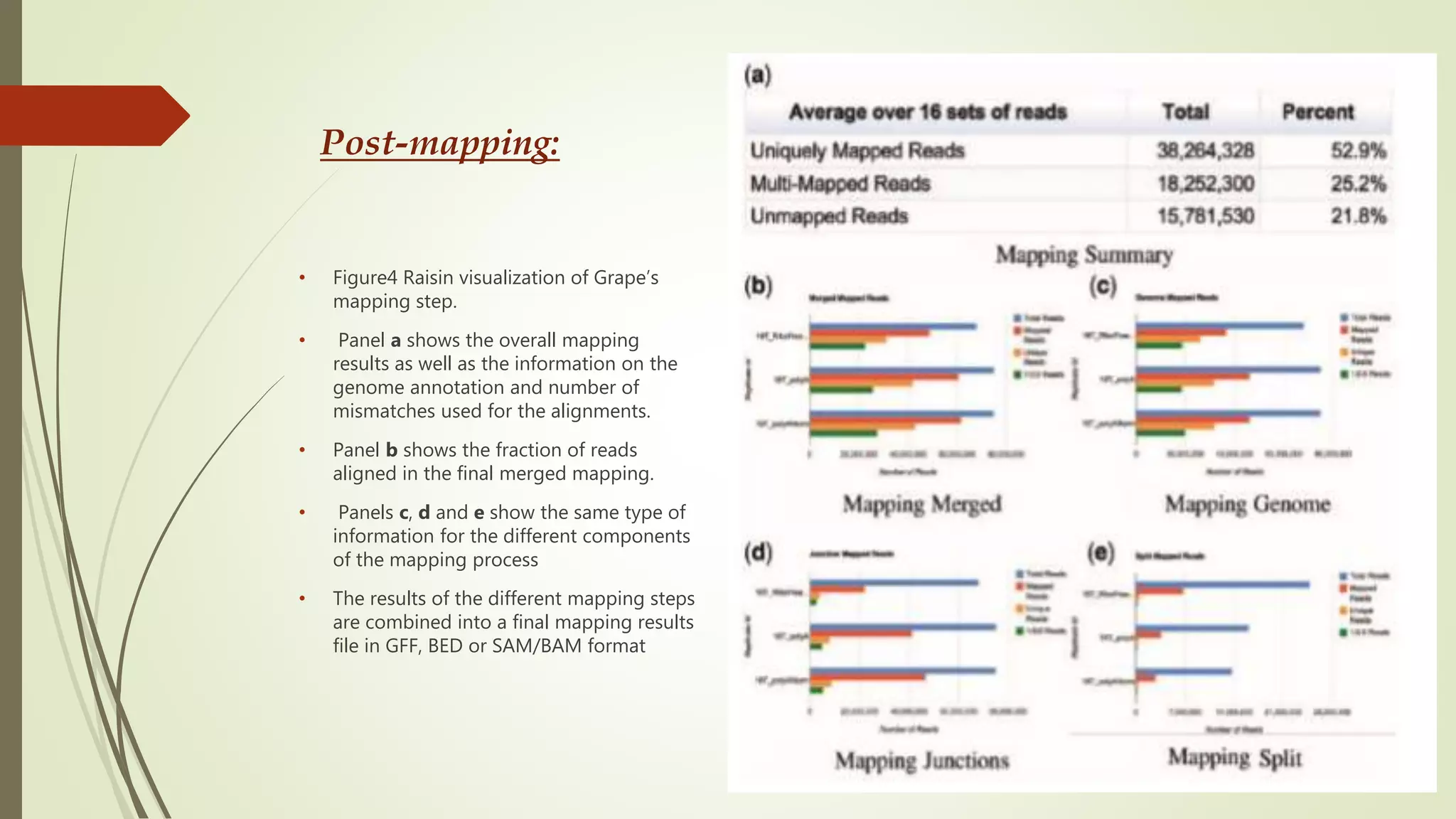
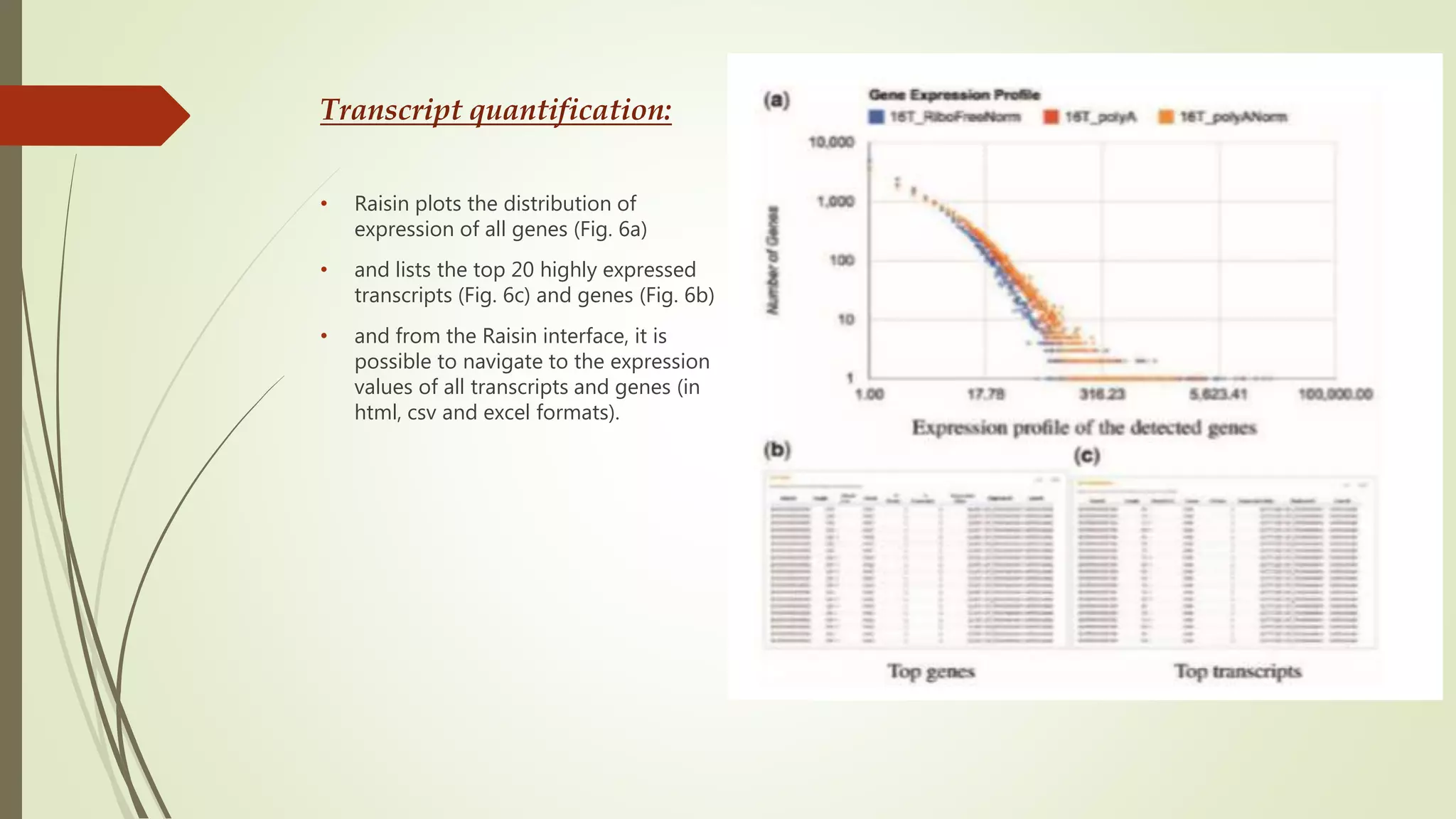
The document discusses the significance of RNA sequencing (RNA-seq) in bioinformatics and the need for efficient analysis tools, highlighting the Grape analysis pipeline's capabilities for managing and visualizing RNA-seq data. It outlines the key steps of Grape's workflow, including preprocessing, mapping, post-mapping, transcript quantification, and discovering novel transcribed elements. Additionally, it addresses RNA-seq analysis using the Bioconductor packages limma, glimma, and edgeR for differential expression analysis and gene set testing.