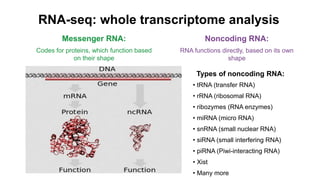
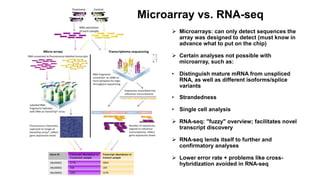
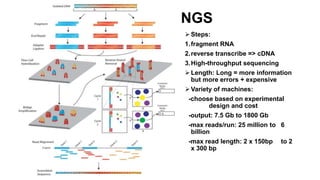
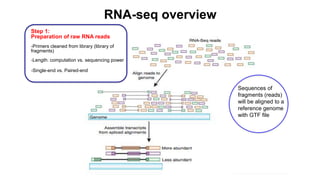
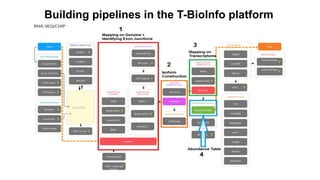
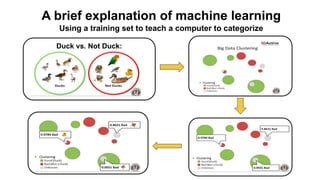
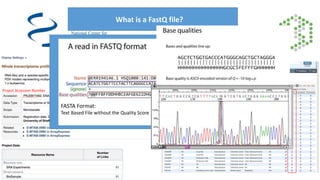
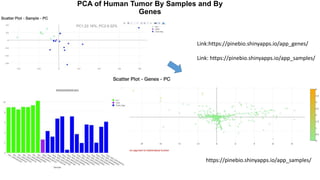
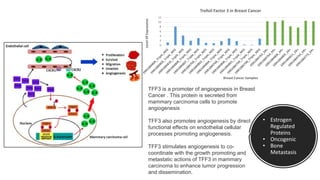
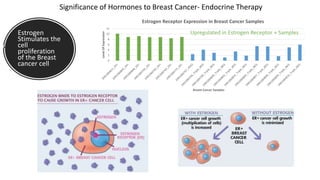
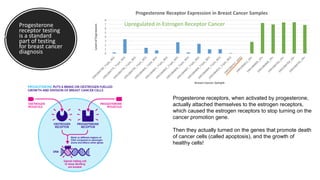
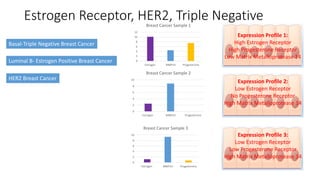
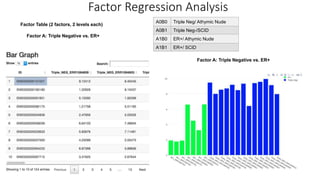
This document provides an overview of RNA-seq analysis using the T-BioInfo platform. It describes analyzing RNA-seq data from breast cancer patient-derived xenograft models to identify differences between cancer subtypes and mouse models. The analysis includes mapping reads, quantifying gene and isoform expression, normalizing data, performing PCA, and identifying biomarker genes for breast cancer subtypes using factor regression analysis. The goal is to gain insights into cancer biology and identify diagnostic or therapeutic targets.