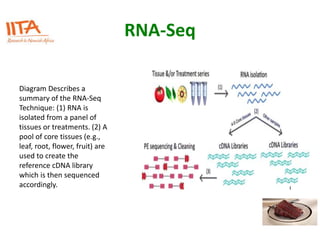
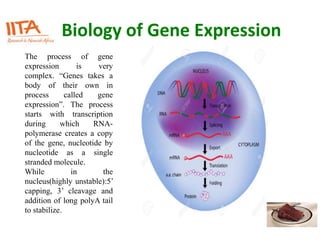
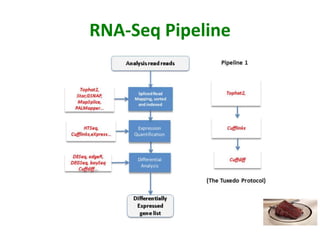
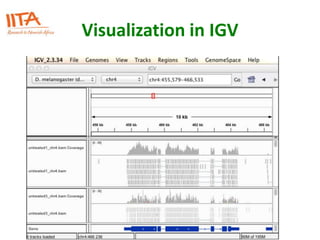
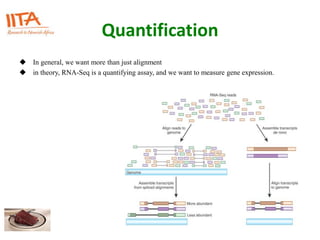
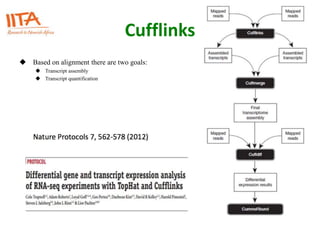
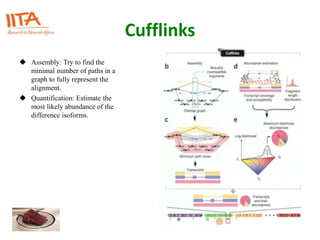
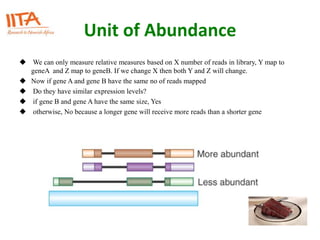
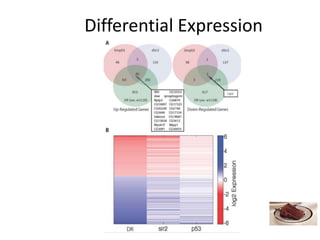
This document outlines the TUXEDO protocol for analyzing RNA-Seq data. It describes the basic steps as: 1) Alignment of RNA-Seq reads to a reference genome or transcriptome using splice-aware aligners like TopHat. 2) Quantification of gene and transcript expression levels using Cufflinks. 3) Quality control checks and 4) Detection of differential expression between conditions using Cuffdiff. Key points covered include RNA-Seq methodology, gene expression, alignment formats, FPKM normalization, and quality control metrics.