Downloaded 33 times
















































































































![18-1140-62 AC 165
227. Ceccarini, C. and Eagle, H. pH as a determinant of cellular growth and contact inhibition. Proc. Natl. Acad. Sci. USA
68, 229–233 (1971).
228. Ceccarini, C. Effect of pH on plating efficiency, serum requirement, and incorporation of radioactive precursors into
human cells. In Vitro 11, 78–86 (1975).
229. Eagle, H. The effect of environmental pH on the growth of normal and malignant cells. J. Cell Physiol. 82,
1–8 (1973).
230. Ham, R. G. Dermal fibroblasts. Methods Cell Biol. 21A, 255–276 (1980).
231. Good, N. E., Wingate, G. D., Winter, W. et al. Hydrogen ion buffers for biological research. Biochemistry 5, 467 (1963).
232. McLimans, W. F. Mass culture of mammalian cells. Meth. Enzymol. 58, 194–211 (1979).
233. Ceccarini, C. and Eagle, H. Induction and reversal of contact inhibition of growth by pH modification. Nature New
Biol. 233, 271–273 (1971).
234. Eagle, H. Buffer combinations for mammalian cell culture. Science 174, 500–503 (1971).
235. Reitzer, L. J., Wice, B. M., Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa
cells. J. Biol. Chem. 254, 2669–2676 (1979).
236. Zielke, H. R., Sumbilla, C. M., Sevdalian, D. A. et al. Lactate: a major product of glutamine metabolism by human
diploid fibroblasts. J. Cell Physiol. 104, 433–441 (1980).
237. Young, D. V., Nakano, E. T. Reduction of lactic acid secreted from SV3T3 cells by a serum factor and biotin and its
relationship to cell growth. Biochem. Biophys. Res. Commun. 93, 1036–1043 (1980).
238. Imamura, T., Crespi, C. L., Bruengraber, H. Utilization of carbohydrate by Madin-Darby canine kidney MDCK cells in
high density suspension of dextran microcarriers. Fed. Proc. 39, 2145 (1980).
239. Cristofalo, V. J. and Kritchevsky, D. Growth and glycolysis in the human diploid cell strain WI-38. Proc. Soc. Exp. Biol.
Med. 118, 1109–1113 (1965).
240. Waymouth, C. Determination and survey of osmolality in culture media, in Tissue Culture: Methods and Applications
(Kruse, P. F. and Patterson, M. K., eds.), Academic Press, New York, pp. 703–709 (1973).
241. Toth, G. M., in Cell Culture and Its Applications (Acton, R.T. and Lynn, J. D., eds.), Academic Press, New York, p. 617 (1977).
242. Griffiths, B. The use of oxidation-reduction potential (ORP) to monitor growth during a cell culture. Dev. Biol. Stand.
55, 113–116 (1984).
243. Fogh, J., ed., Contamination in Tissue Culture, Academic Press, New York (1973).
244. Litwin, J. The effect of antibiotics on the growth and longevity of human diploid fibroblasts in vitro. Acta Pathol.
Microbiol. Scand. [B] Microbiol. Immunol. 81, 593–605 (1973).
245. Goetz, I. E., Moklebust, R., Warren, C. J. Effects of some antibiotics on the growth of human diploid skin fibroblasts in
cell culture. In Vitro 15, 114–119 (1979).
246. Schwarze, P. E. and Seglen, P. O. Effects of antibiotics on protein synthesis and degradation in primary cultures of rat
hepatocytes. In Vitro 17, 71–76 (1981).
247. Aubert-Tulkens, G., van Hoof, F., Tulkens, P. D. Gentamicin-induced lysosomal phospholipidosis in cultured rat
fibroblasts. Quantitative ultrastructural and biochemical study. Lab. Invest 40, 481–491 (1979).
248. Stanbridge, E. Mycoplasmas and cell cultures. Bacteriol. Rev. 35, 206–227 (1971).
249. Chen T. R. In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp.
Cell Res. 104, 255–262 (1977).
250. Hessling, J. J., Miller, S. E., Levy, N. L. A direct comparison of procedures for the detection of mycoplasma in tissue
culture. J. Immunol. Methods 38, 315–324 (1980).
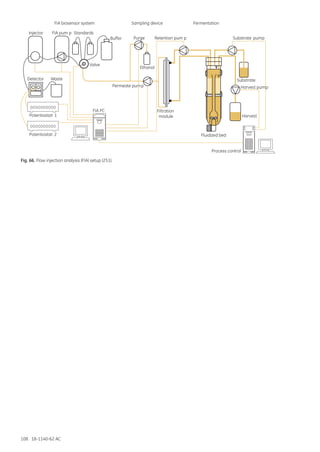
251. Loibner, A. P., Zach, N., Doblhoff-Dier, O. et al. On-line glucose control of animal cell cultures in fluidized beds, in
Animal Cell Technology: Products of Today, Prospects for Tomorrow (Spier, R. E., Griffiths, J. B., Bertold, W., eds.),
Butterworth Heinemann, pp. 372–375. (1994).
252. van der Pol, J. J., Spohn, U., Eberhardt, R. et al. On-line monitoring of an animal cell culture with multi-channel flow
injection analysis. J. Biotechnol. 37, 253–264 (1994).
253. Clark, J., Hirtenstein, M., Gebb, C. Critical parameters in the microcarrier culture of animal cells. Dev. Biol. Stand. 46,
117–124 (1980).
254. van Wezel, A., Rijksinstituut voor de Volksgezondheid, P.O.B. 1, NL-3720 BA Bilthoven, The Netherlands.
255. Holley, R. W. Control of growth of kidney epithelial cells in culture. Hormones and Cell Culture, Cold Spring Harbor
Conferences on Cell Proliferation 6, 455–459 (1979).
256. Cooper, P. D., Wilson, J. N., Burt, A. M. The bulk growth of animal cells in continuous suspension culture.
J. Gen. Microbiol. 21, 702–720 (1959).](https://image.slidesharecdn.com/3veadvo4q6iqer3waeot-140626002839-phpapp02/85/culture-cellular-167-320.jpg)





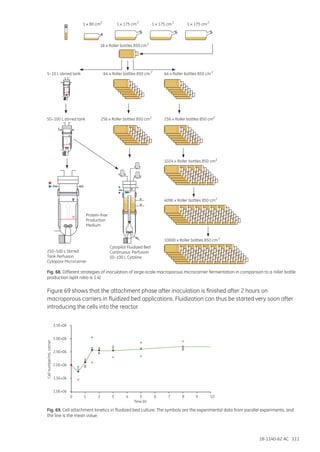
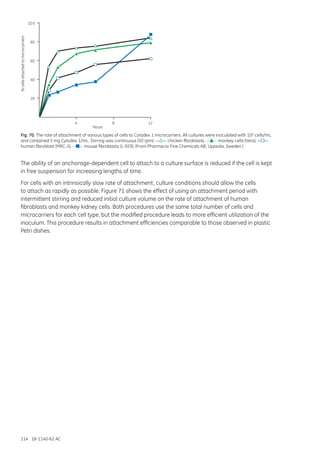
This document provides an overview of microcarrier cell culture principles and methods. Microcarriers allow for increased surface area for cell attachment and growth compared to traditional cell culture methods. They enable higher cell densities and product yields by separating cells from culture medium, improving process control and scalability. Various microcarrier types made from different materials are available for culturing different cell types like mammalian, insect and plant cells. Microcarriers are used to produce vaccines, proteins, antibodies and more in small-scale equipment like spinners and roller bottles as well as large-scale bioreactors like stirred tanks, packed beds and fluidized beds. Careful selection of microcarrier properties, culture conditions and control strategies are required to optimize microcarrier cell culture











































































































