Downloaded 98 times





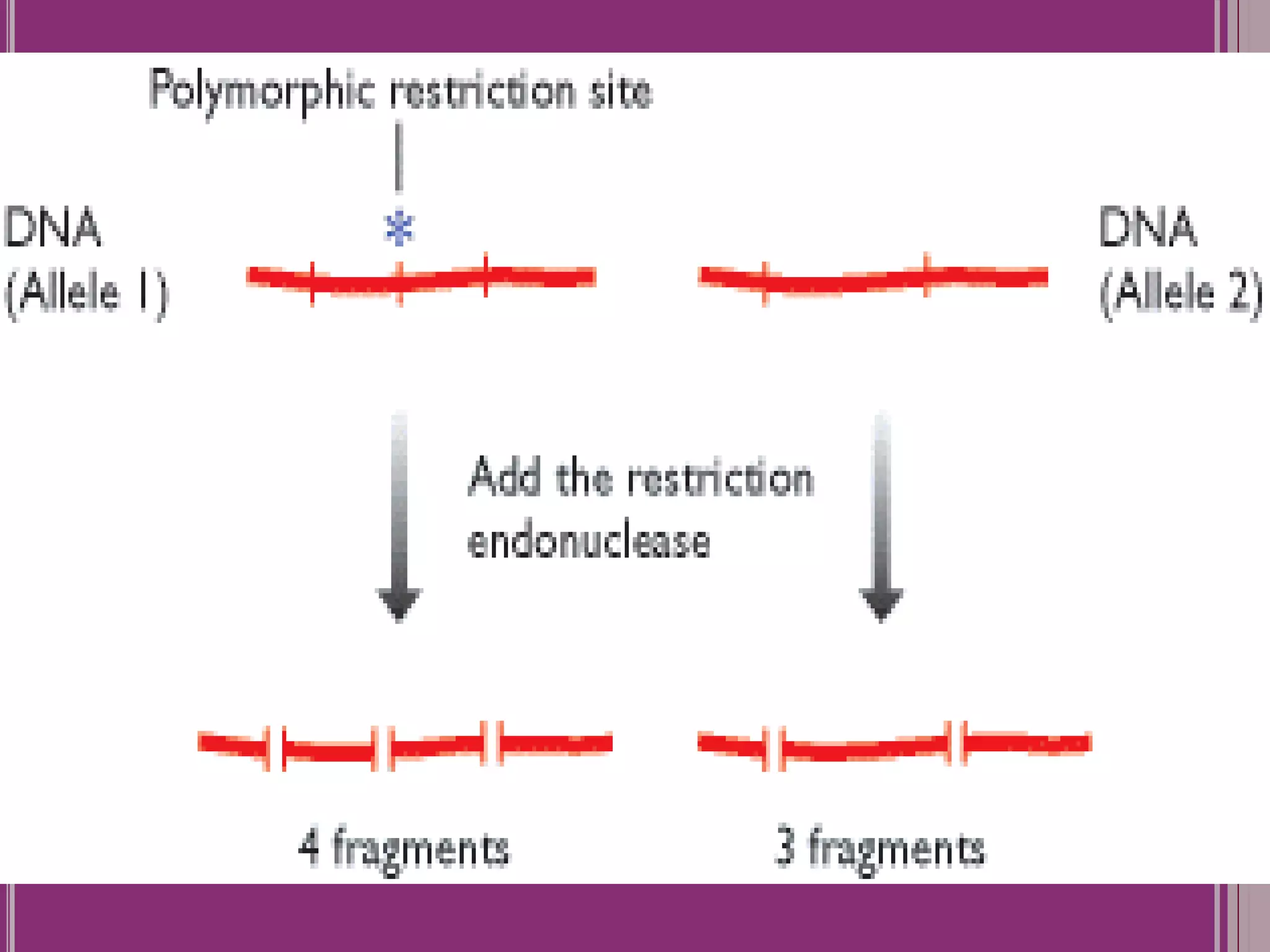






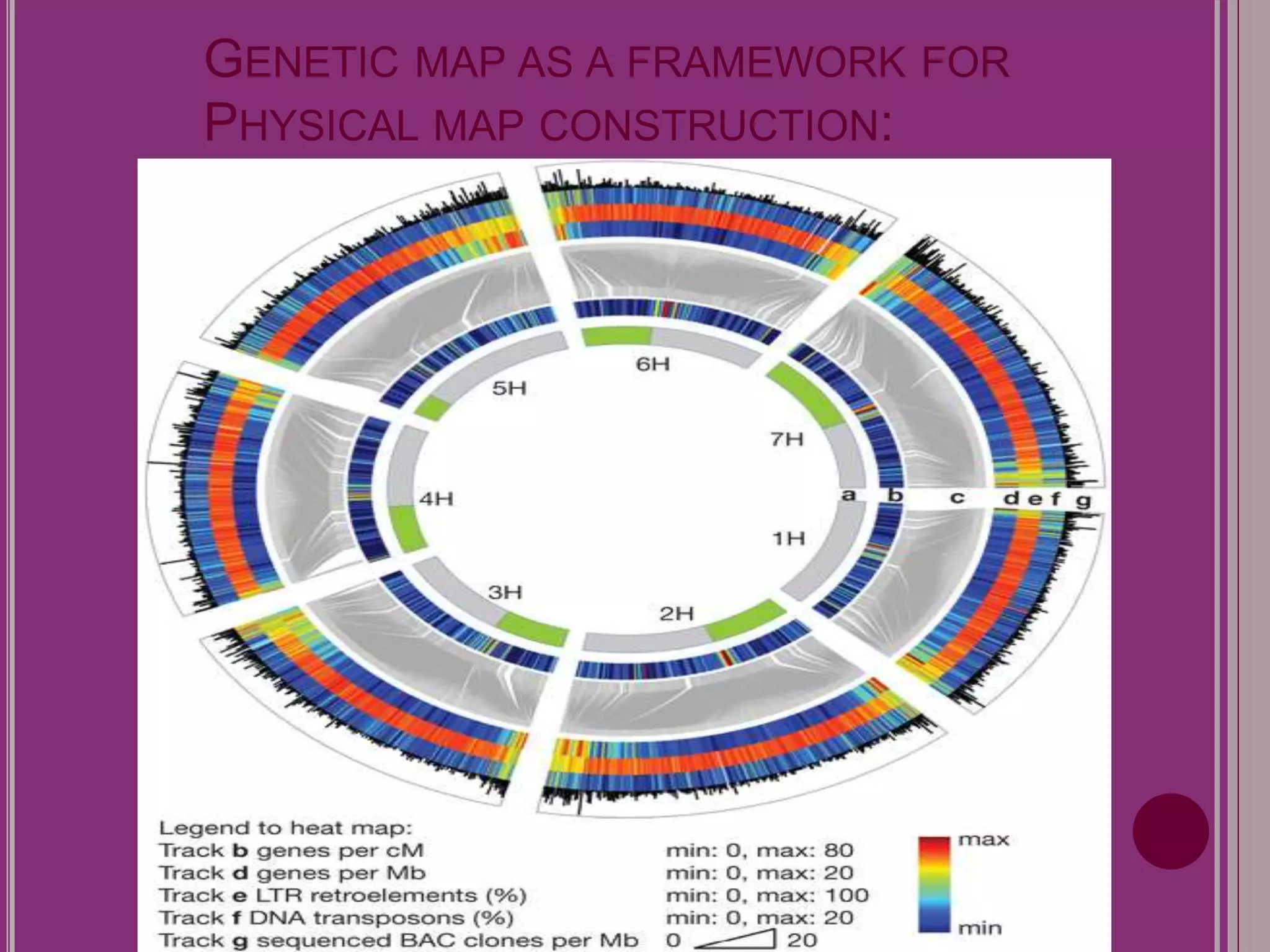

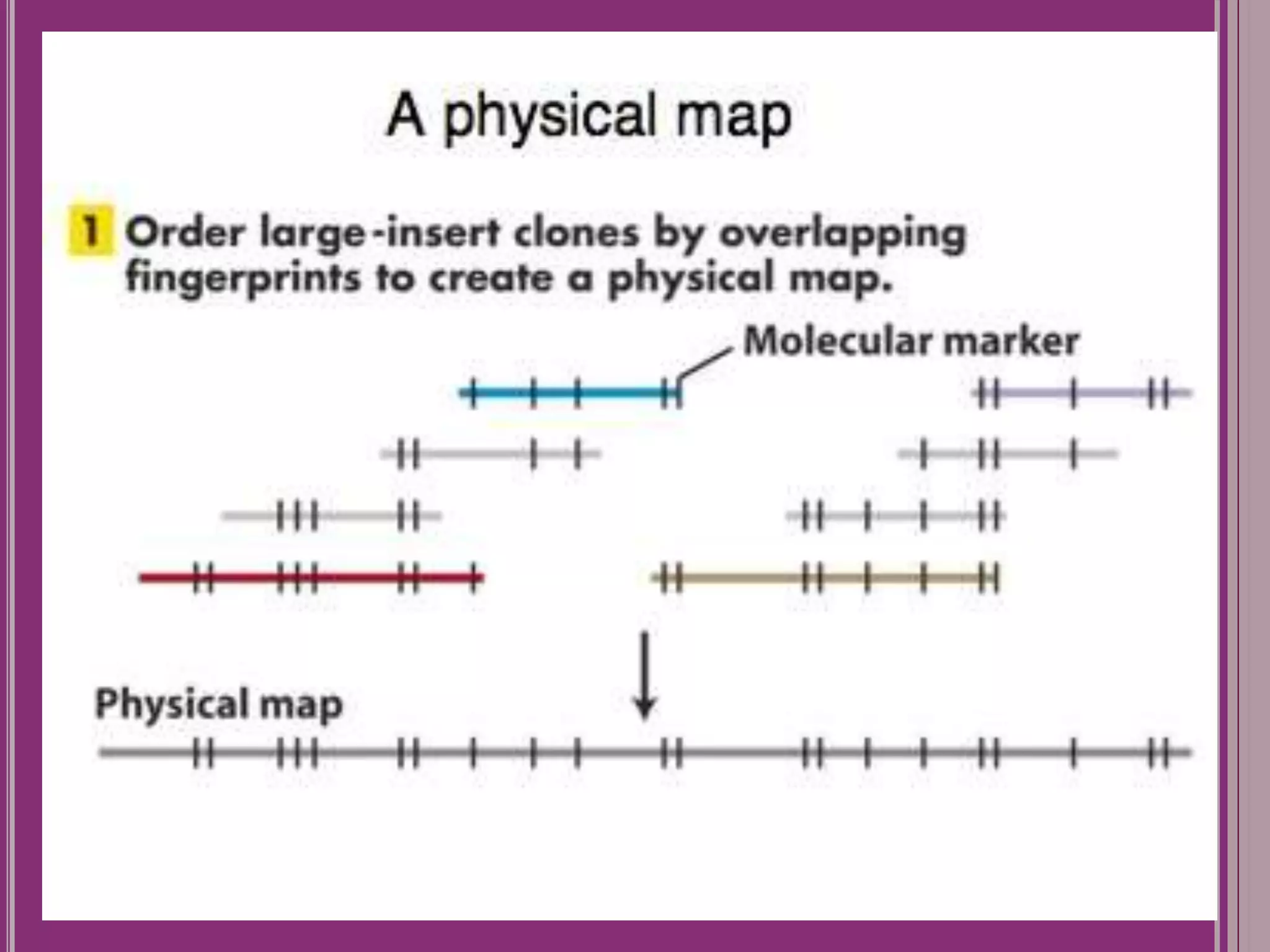



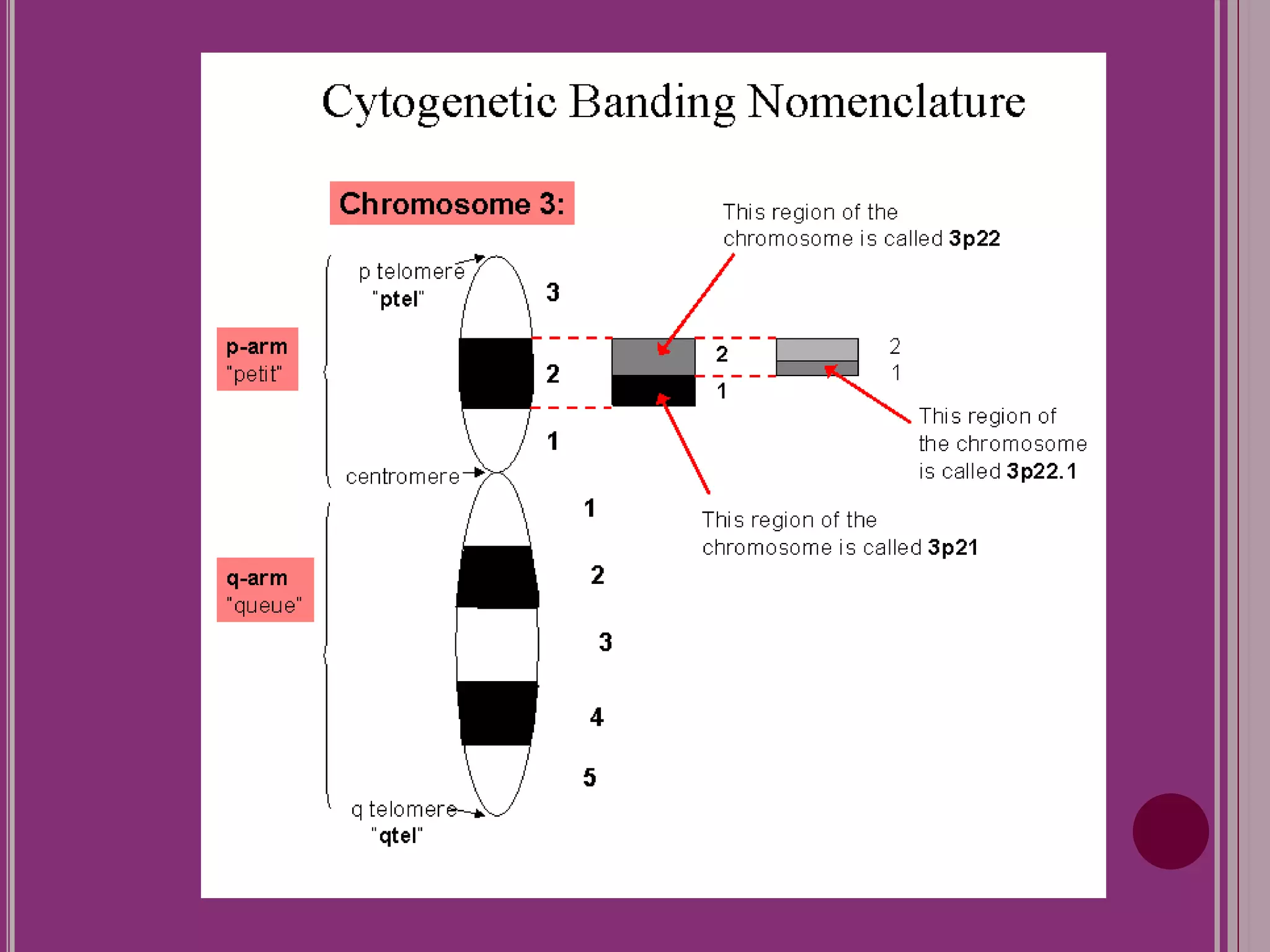



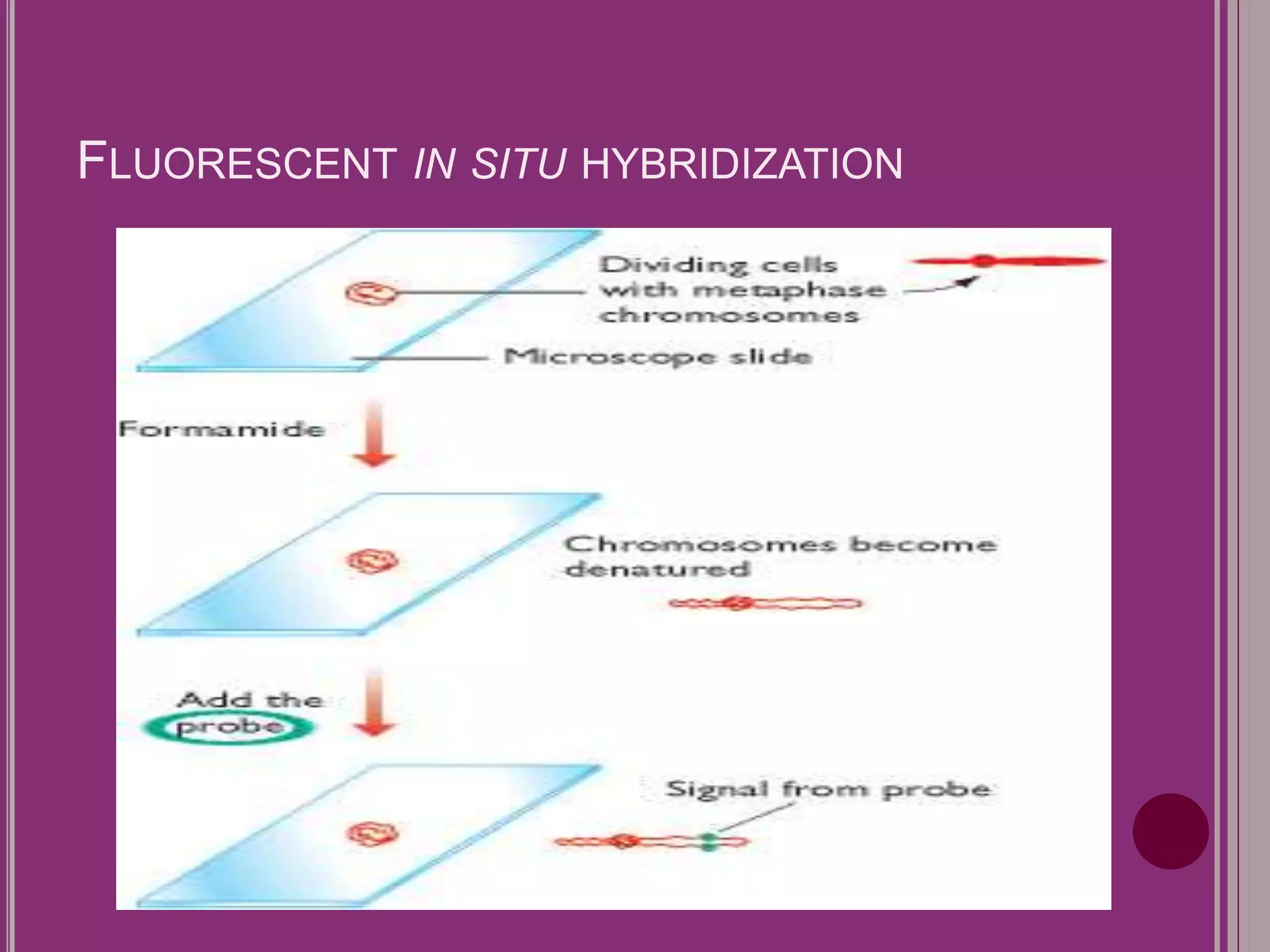


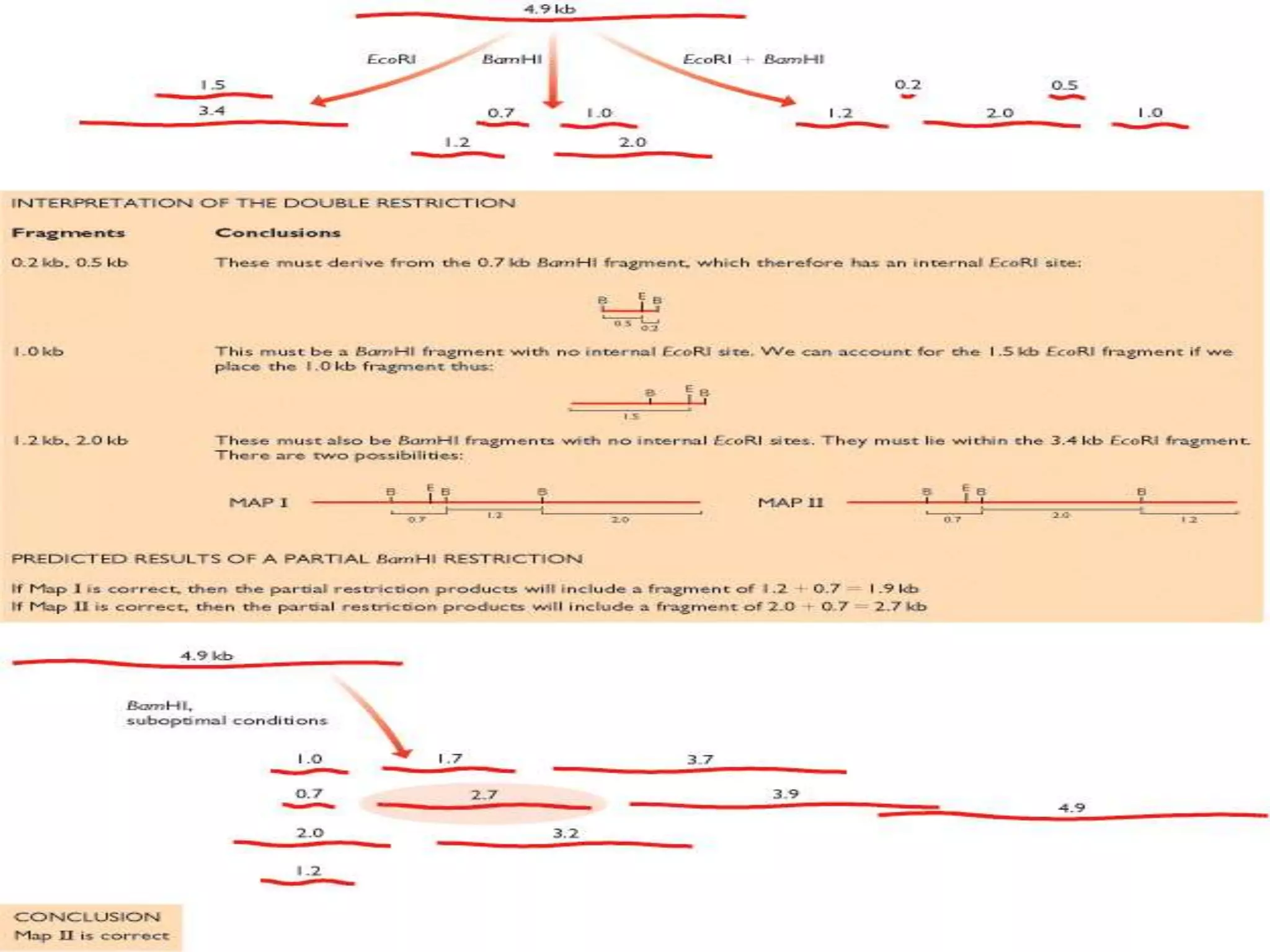

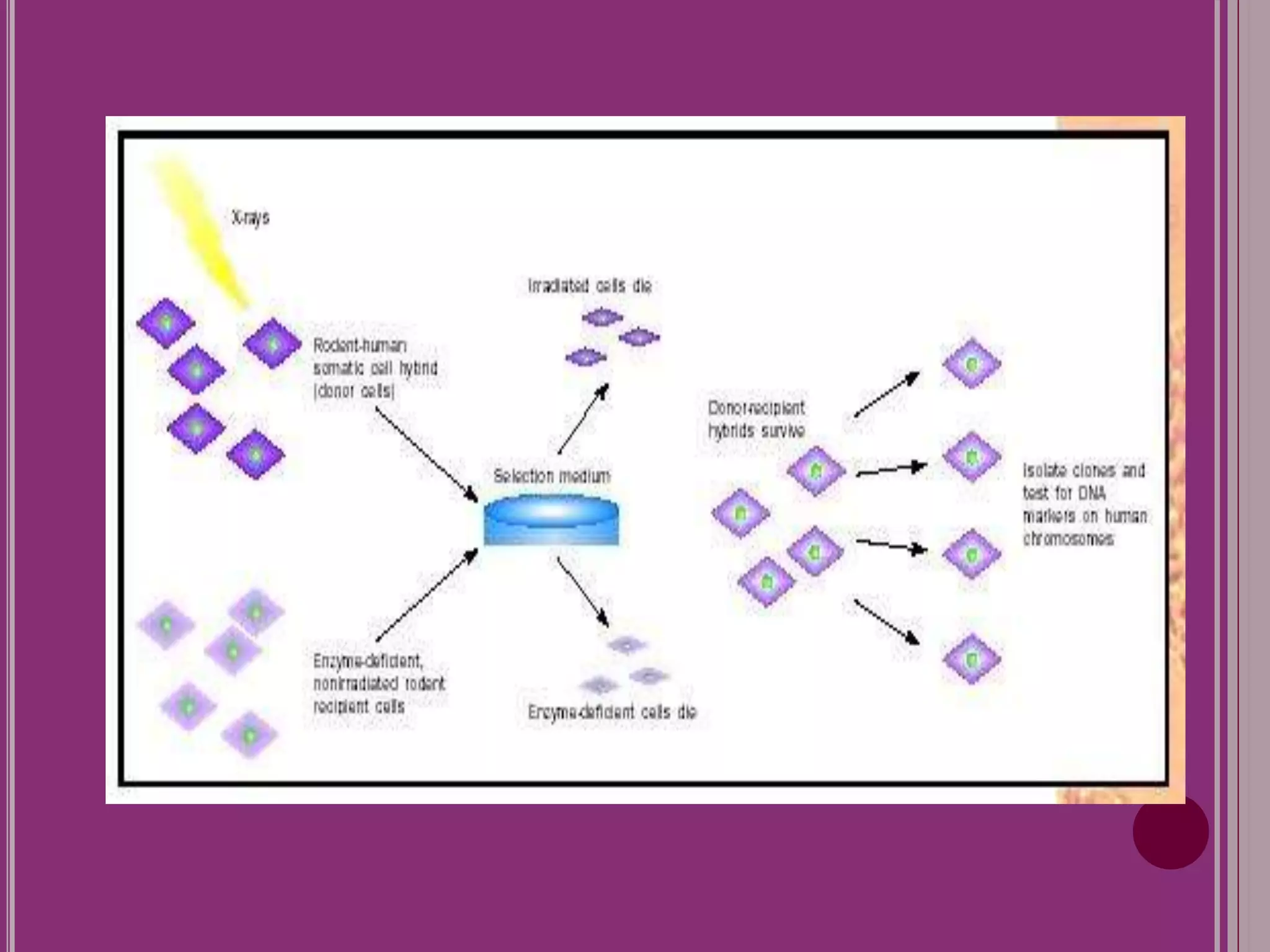

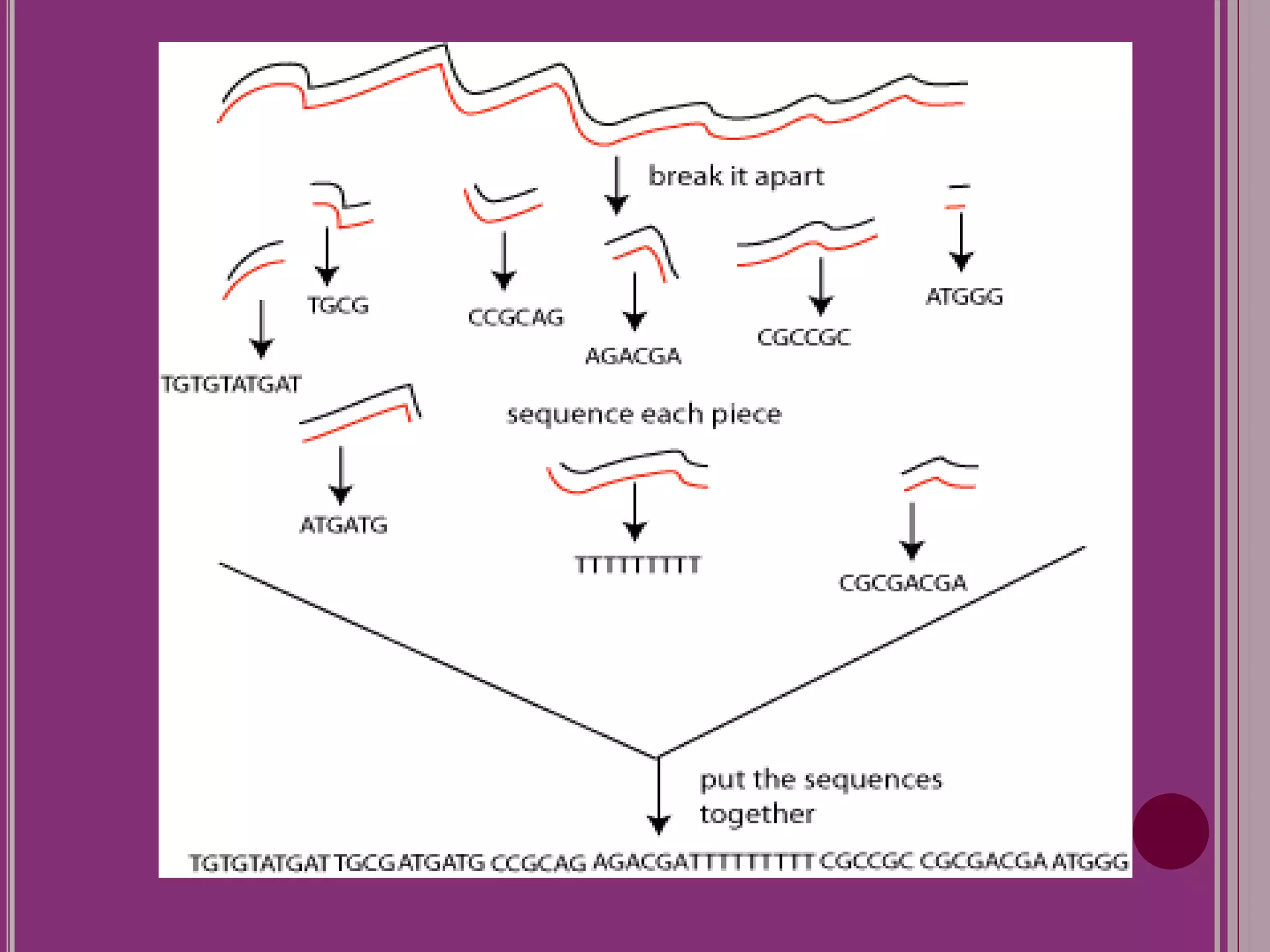


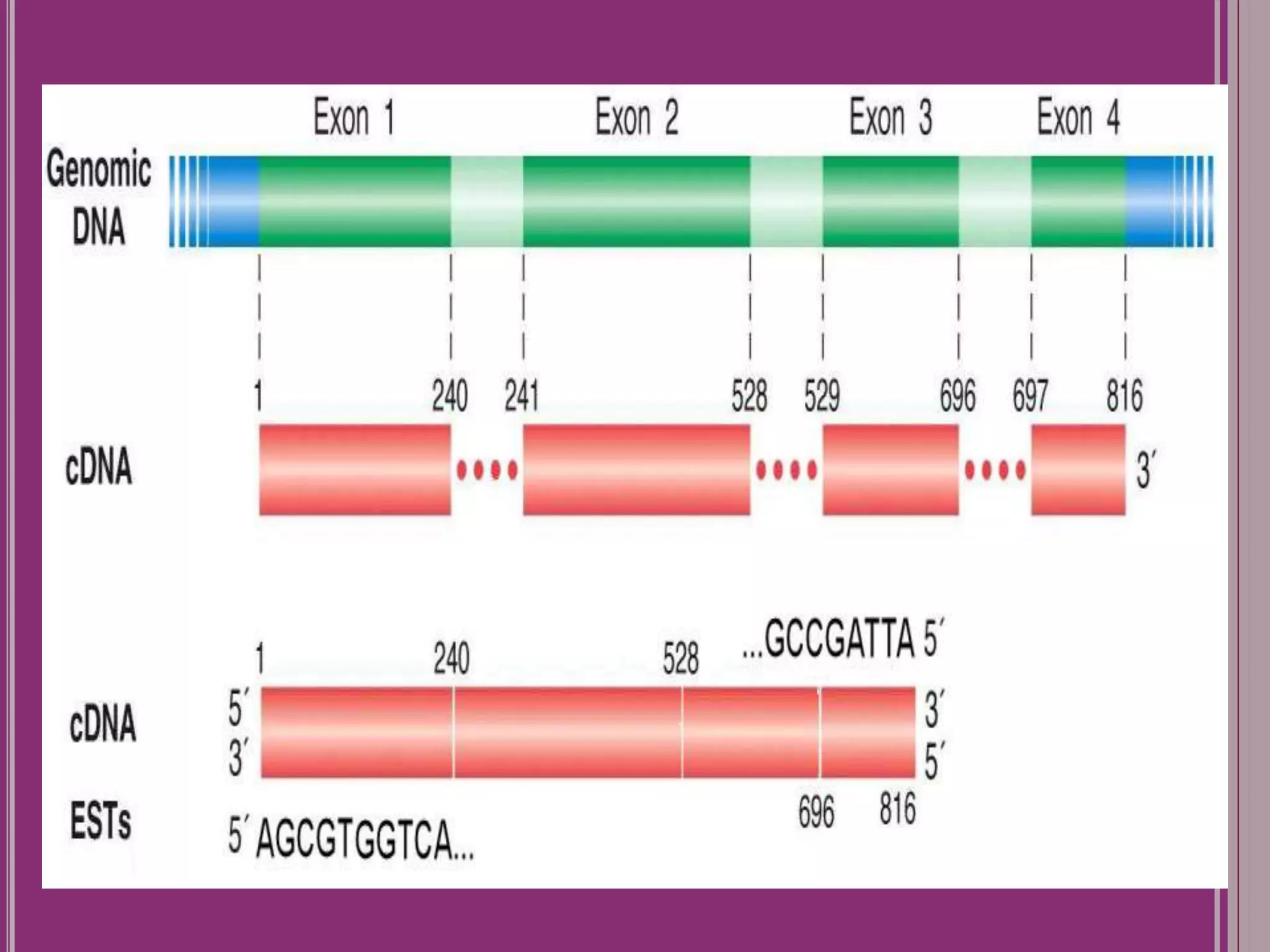

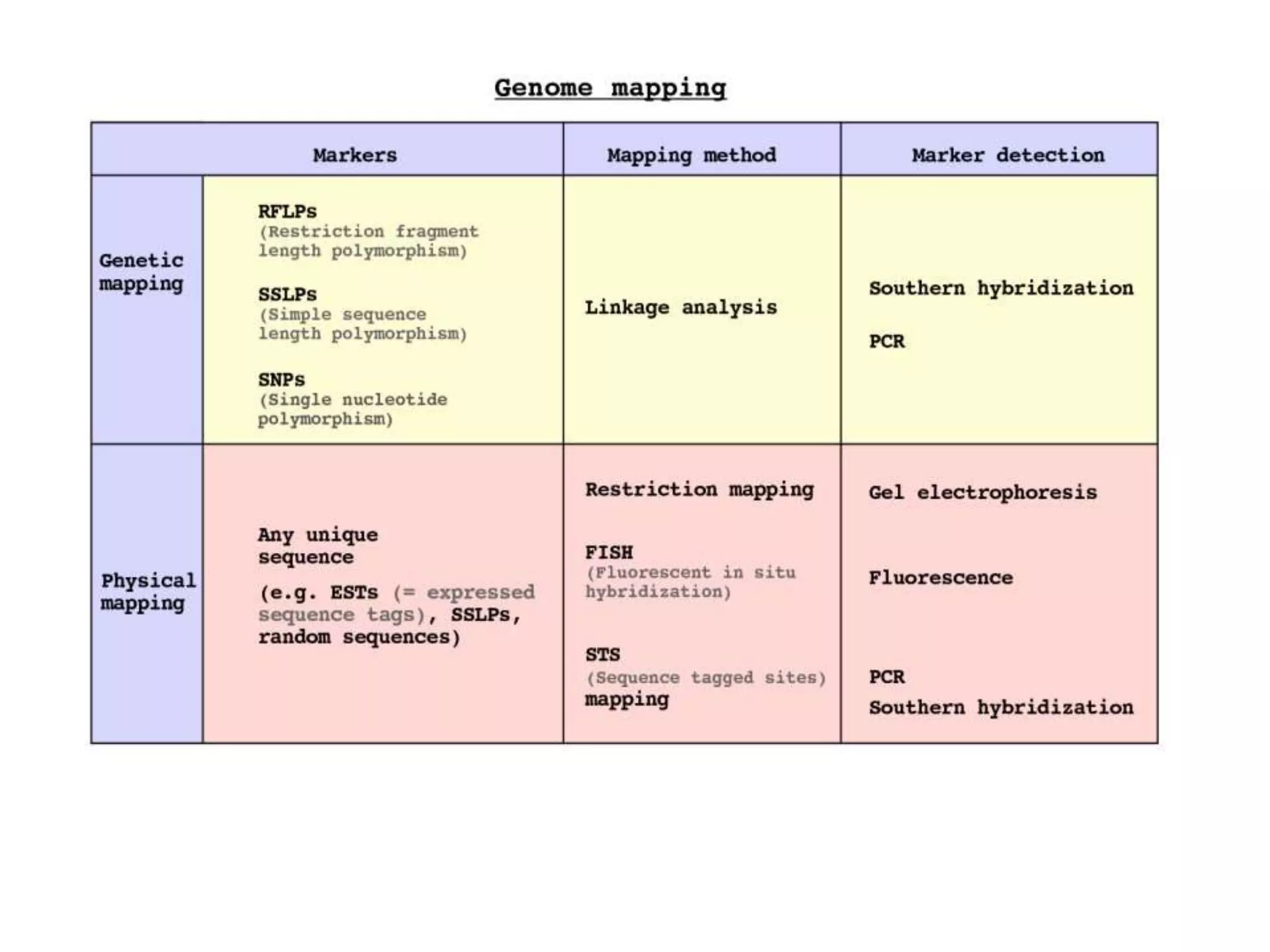









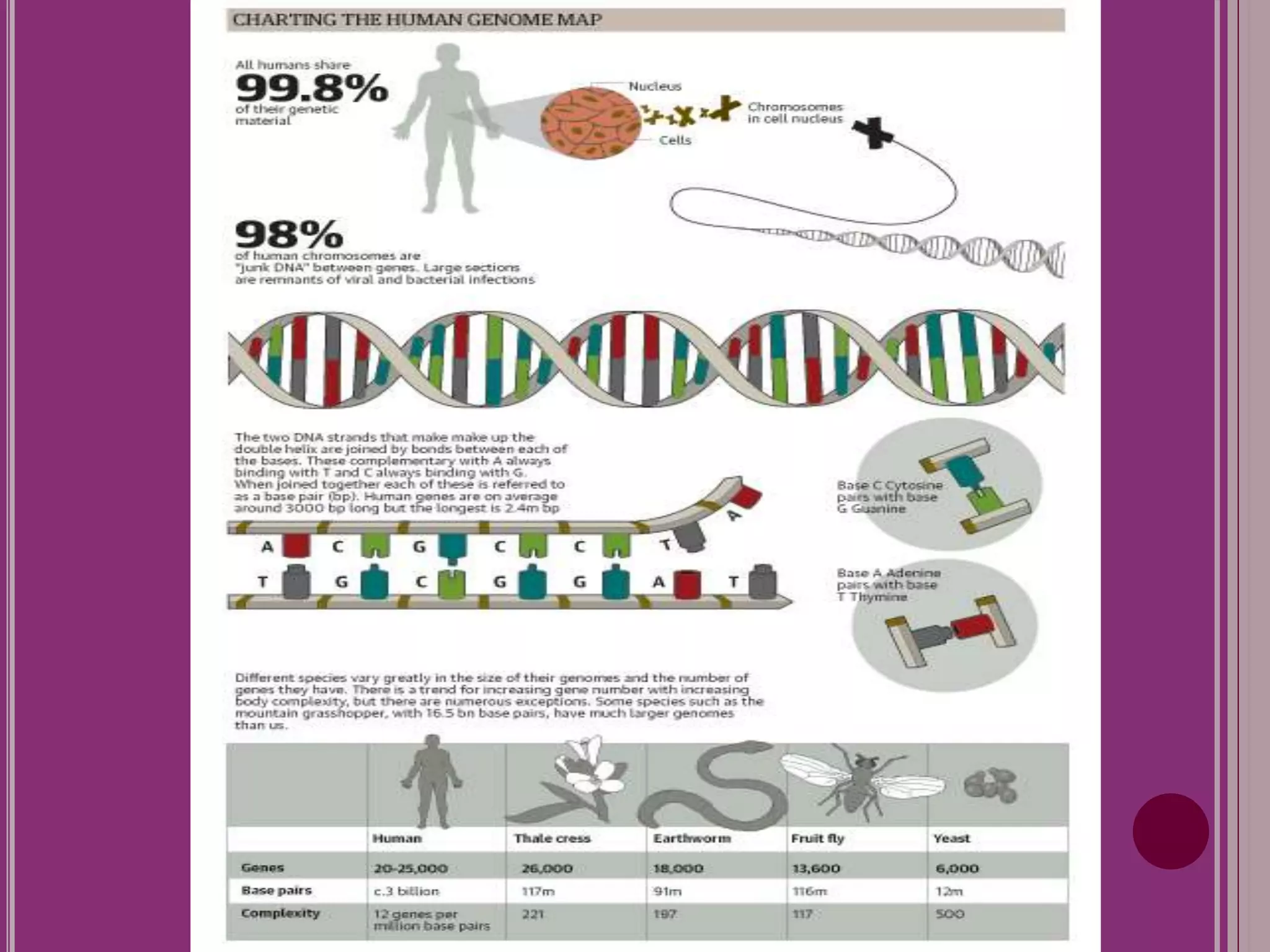


The document outlines the construction of a human gene map using integration from genetic maps to physical maps and sequence maps, focusing on various DNA markers such as RFLPs, SSLPs, and SNPs. It describes the methods used to measure genetic linkage and physical distances on chromosomes, highlighting the importance of technologies like FISH and sequencing methods such as the clone contig and whole genome shotgun approaches. The ultimate goal is to fully sequence the human genome to aid in understanding genes and developing treatments for diseases.



































































