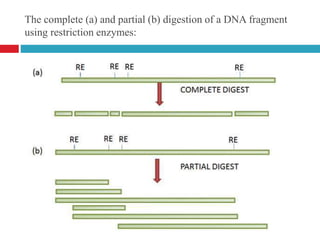
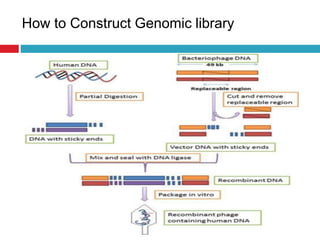
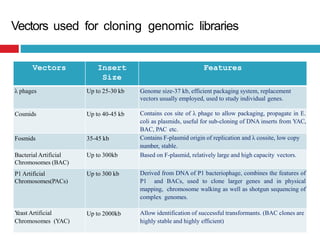
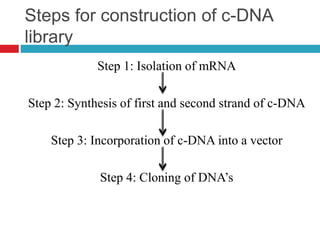
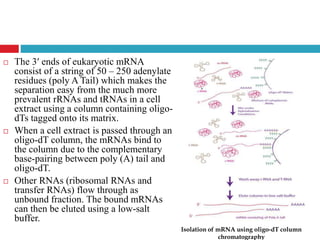
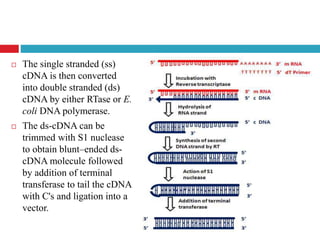
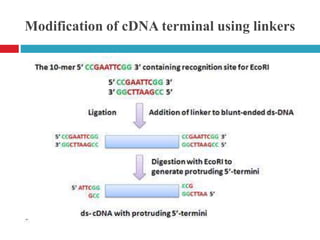
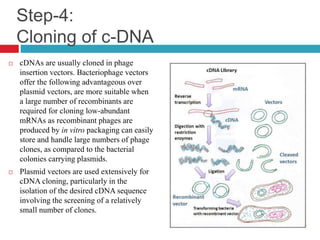
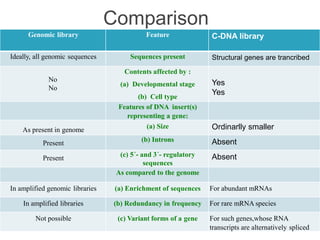
Genomic and cDNA libraries allow for the representation of genomic sequences as multiple small fragments. A genomic library contains fragments from all DNA sources, including coding and non-coding regions. A cDNA library contains only expressed coding sequences, as it is synthesized from mRNA. The process involves isolating mRNA, synthesizing cDNA, incorporating it into a vector, and cloning the fragments. Libraries are important tools for studying genomes, genes, and gene expression.






























































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)