Downloaded 38 times





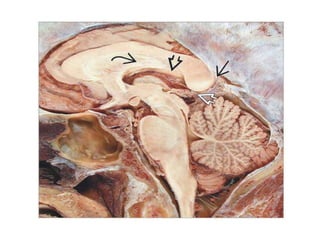
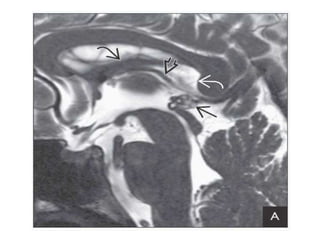



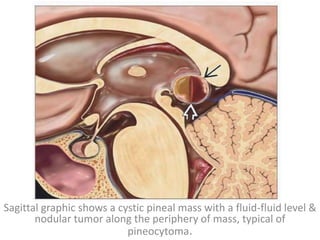
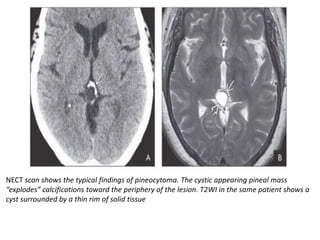


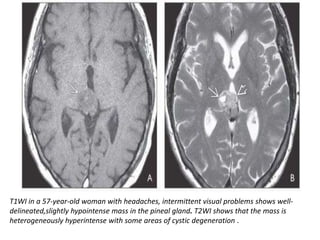
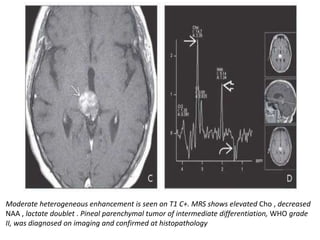


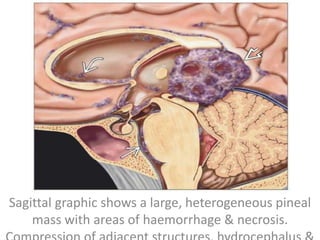
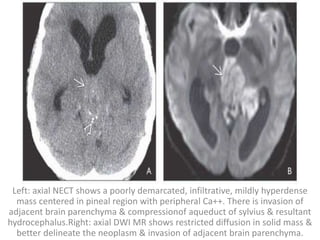
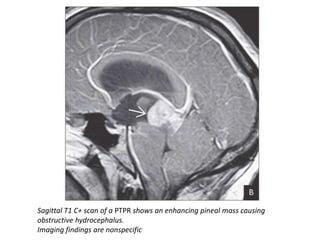


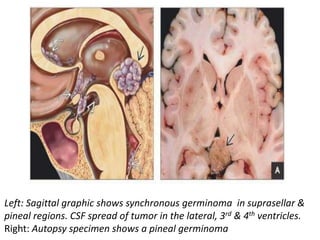
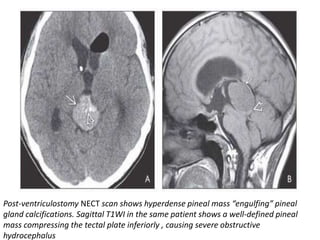
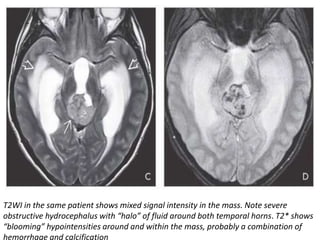
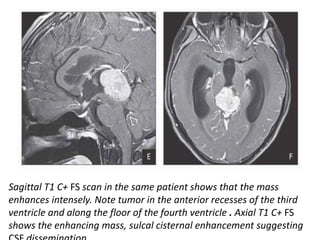
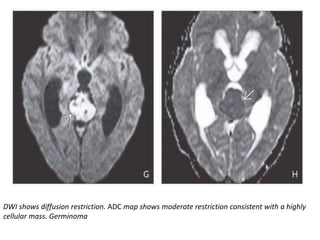


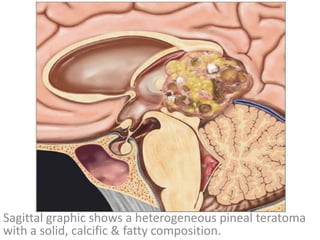
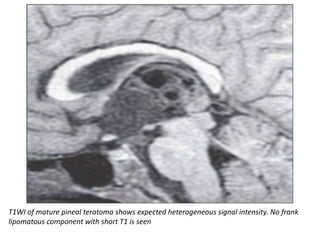









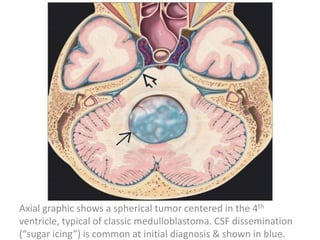

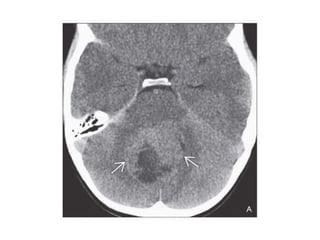
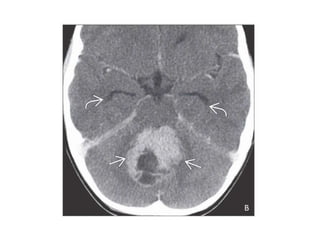
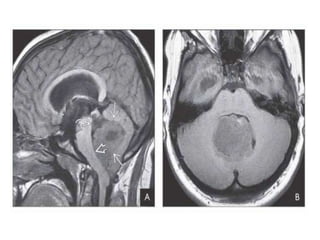
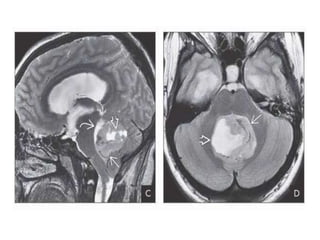
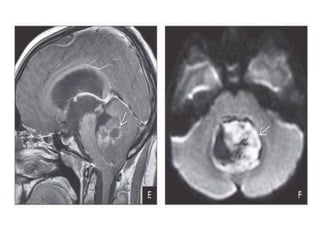


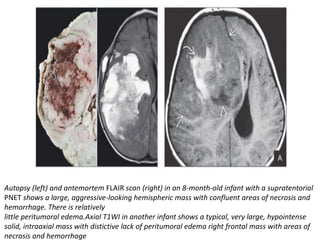
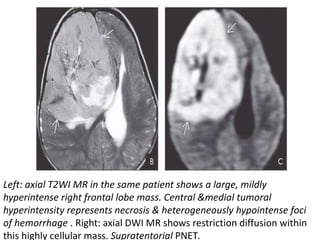
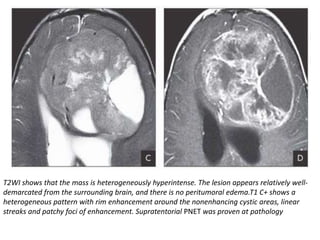






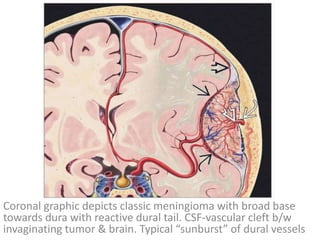



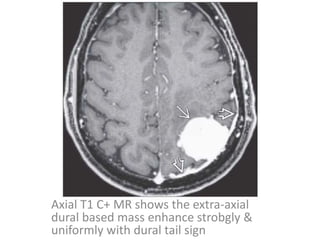
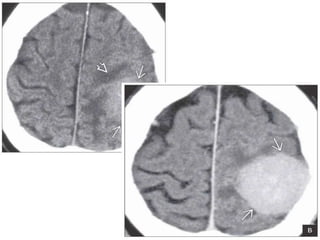
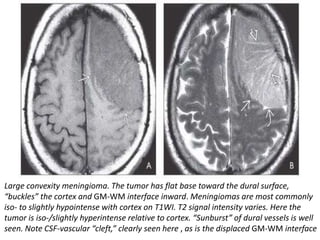
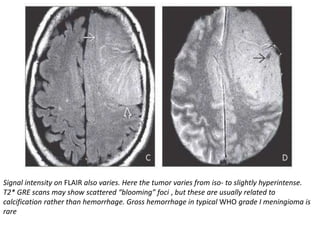
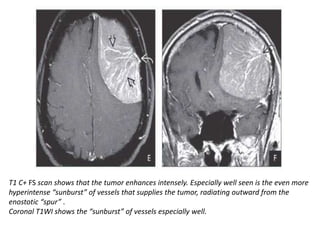

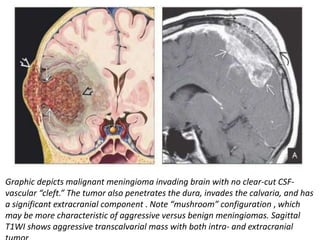





The document provides an extensive overview of various types of brain tumors, particularly those affecting the pineal region and neuroepithelial tissues, including their classification, imaging characteristics, and differential diagnosis. It highlights common tumors such as pineocytoma, pineoblastoma, and germinomas, detailing their presentation, imaging findings, and clinical significance. Additionally, the document discusses embryonal tumors like medulloblastoma and neuroblastic tumors, emphasizing their aggressive nature and classification according to the World Health Organization.
















































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)




