Download to read offline



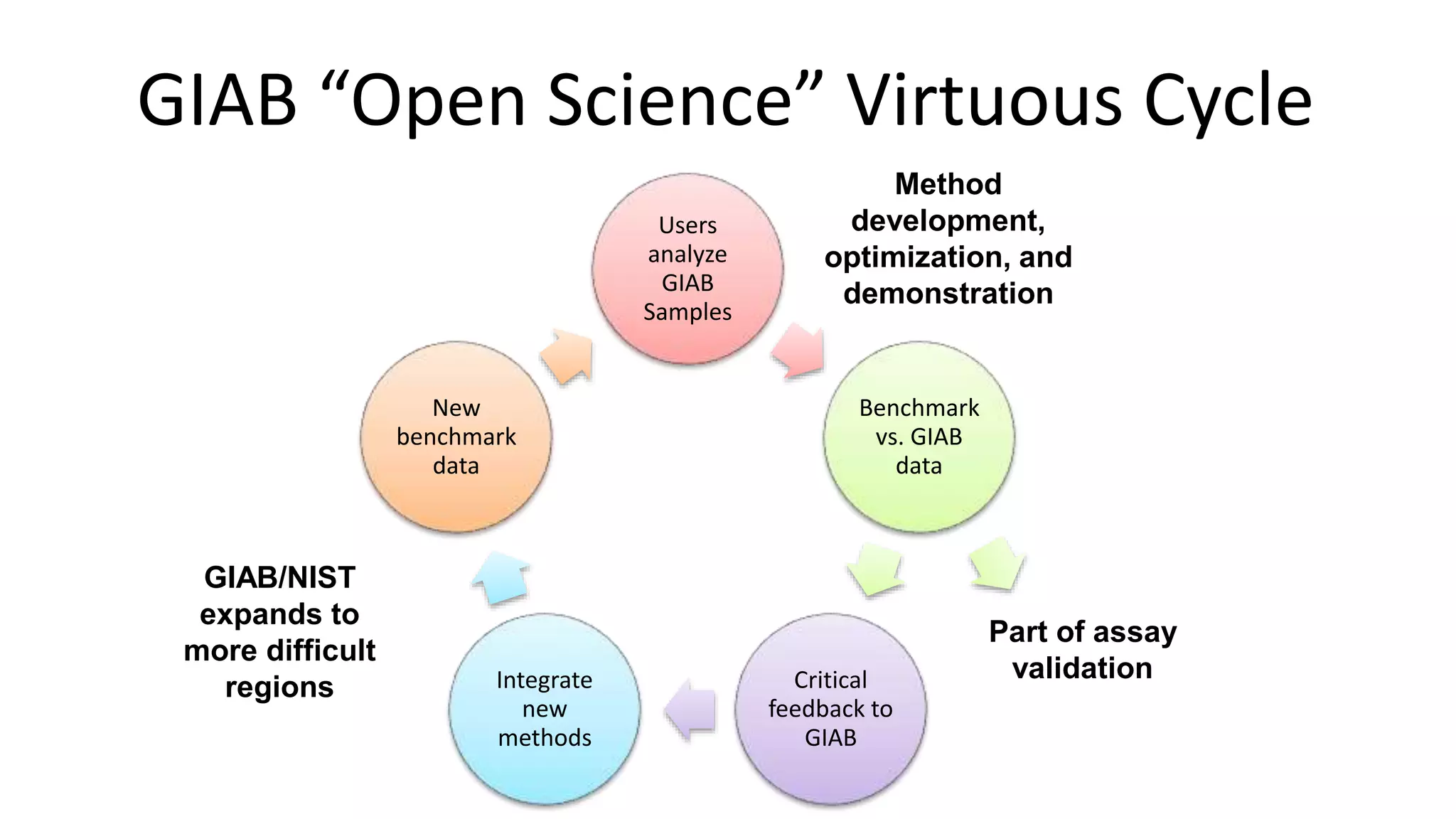
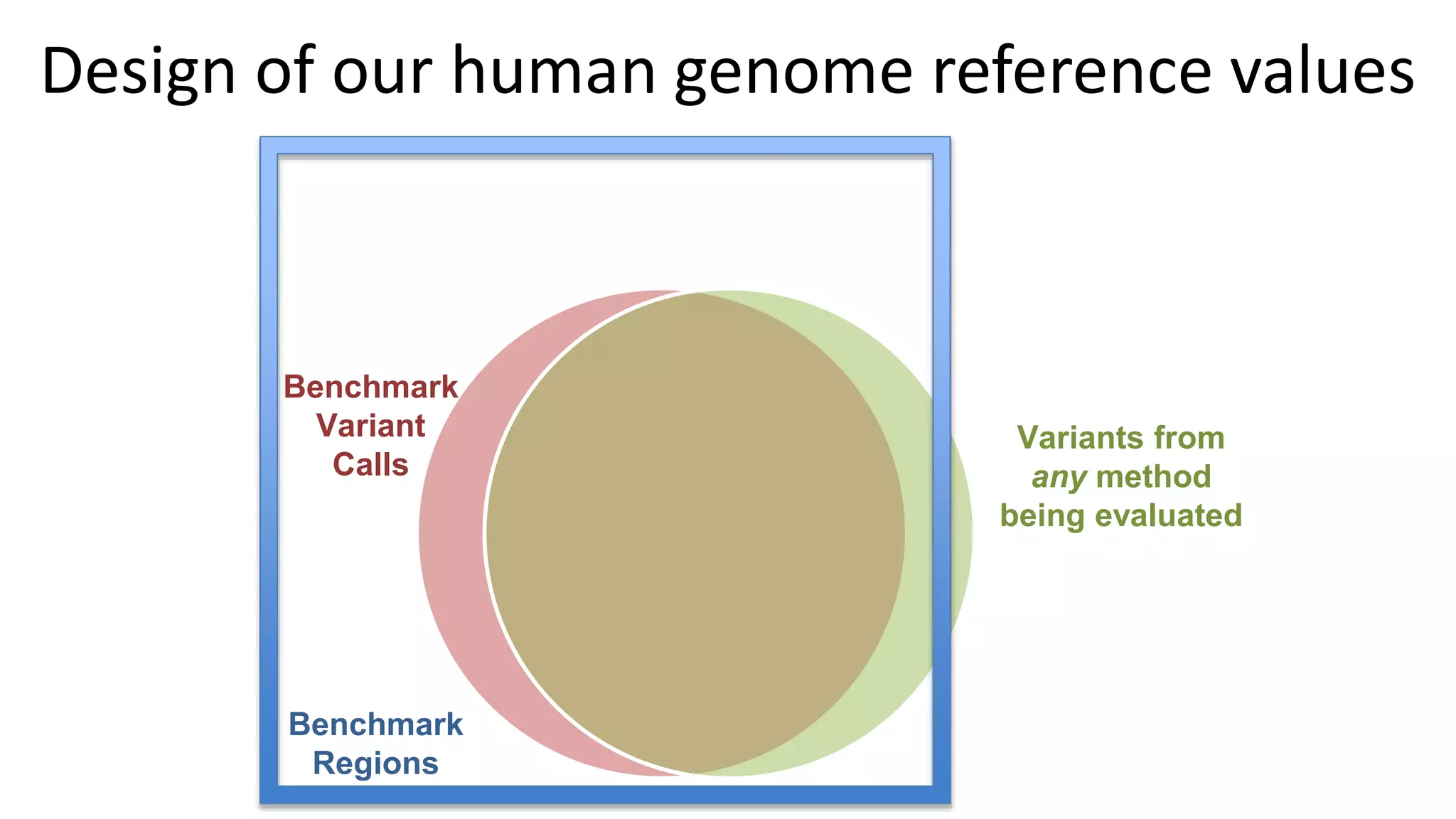
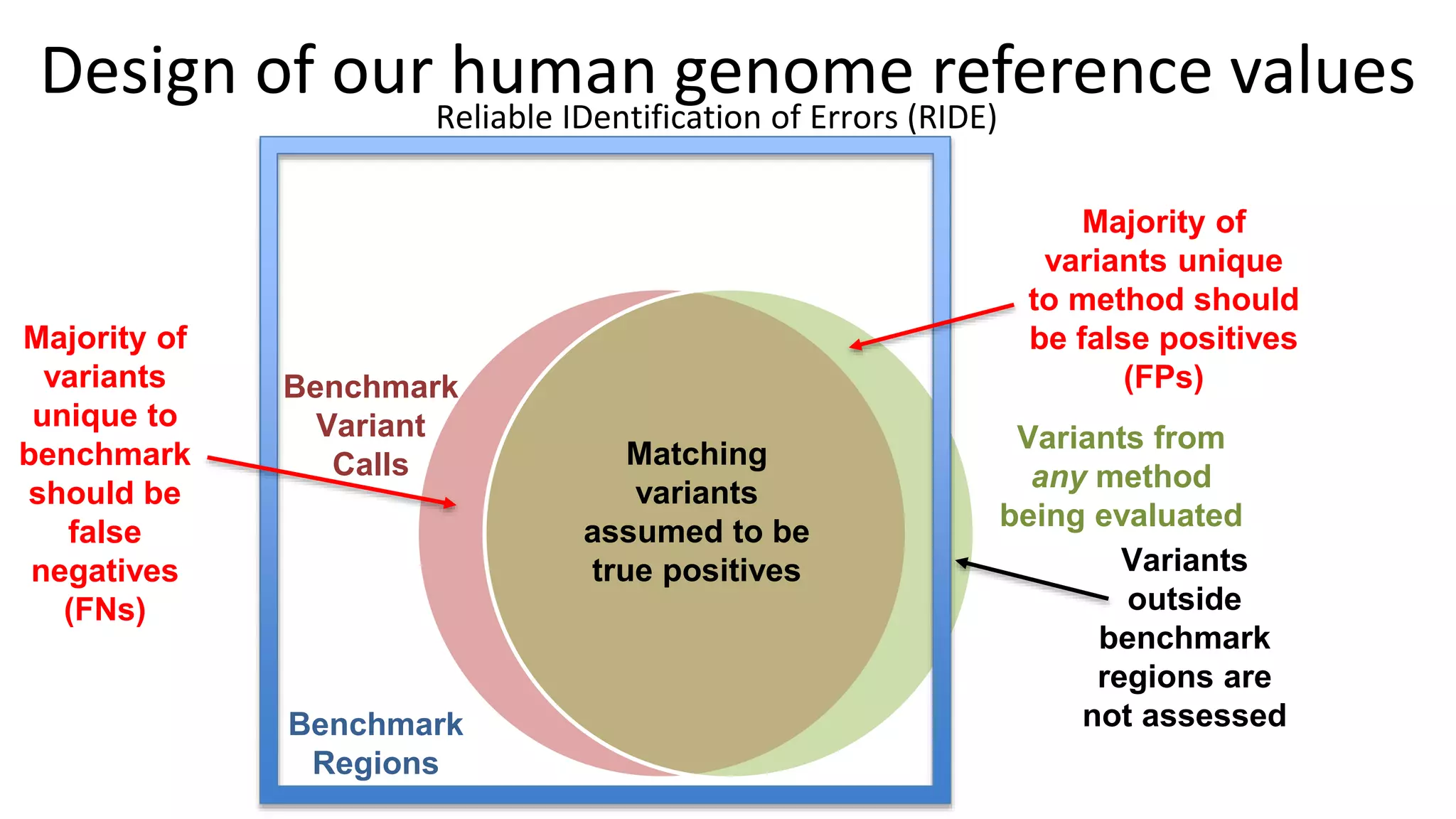


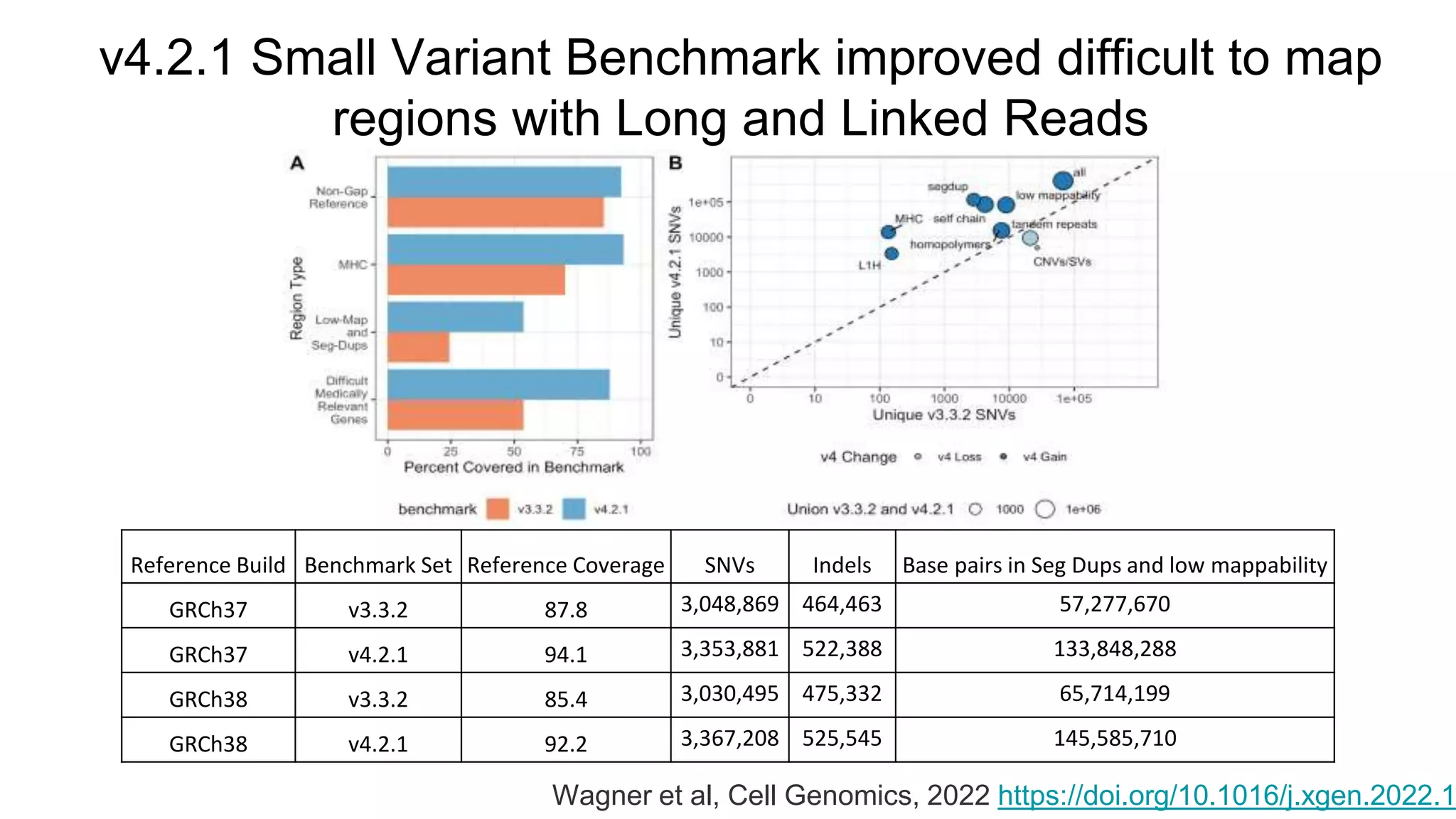

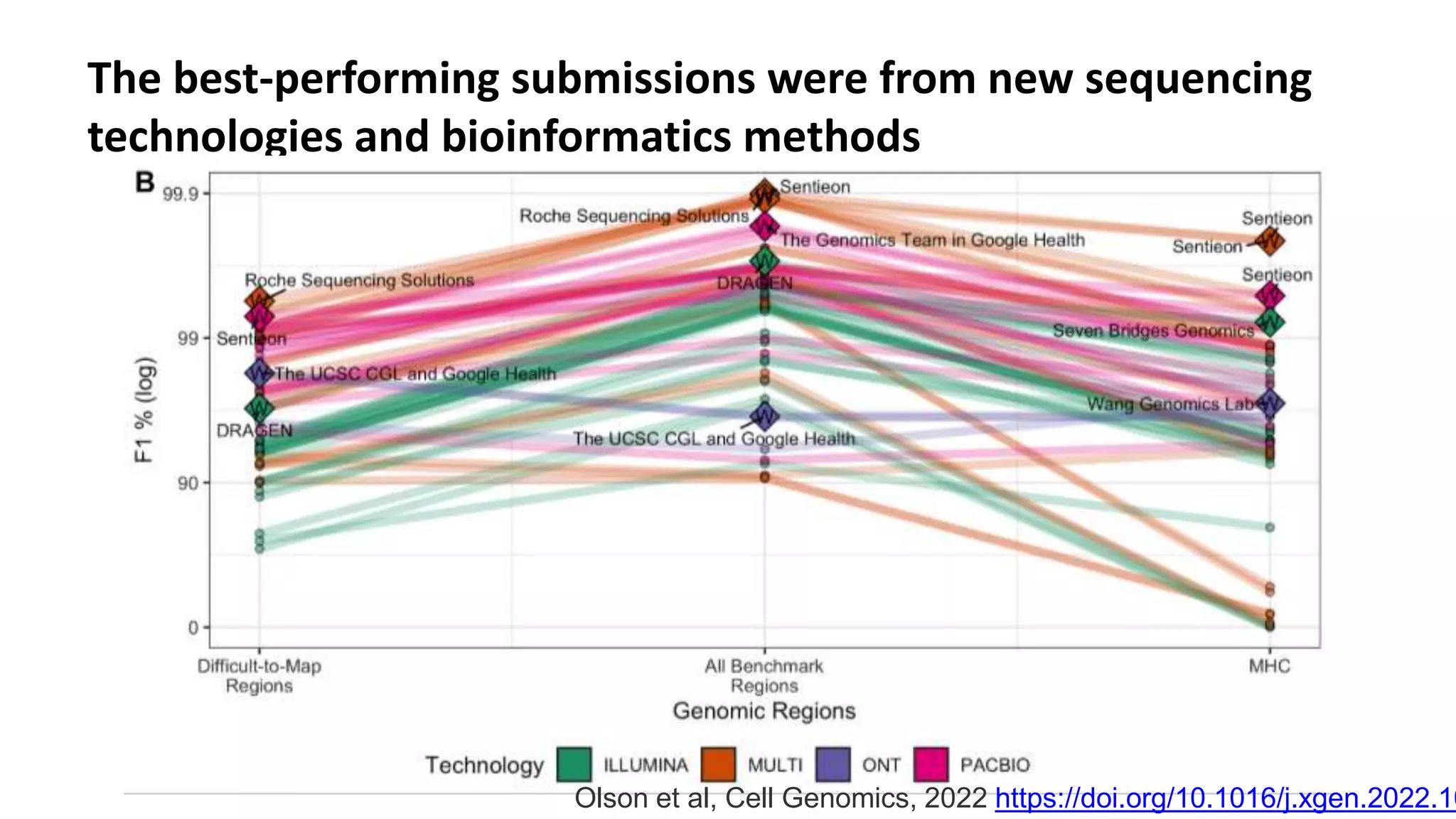
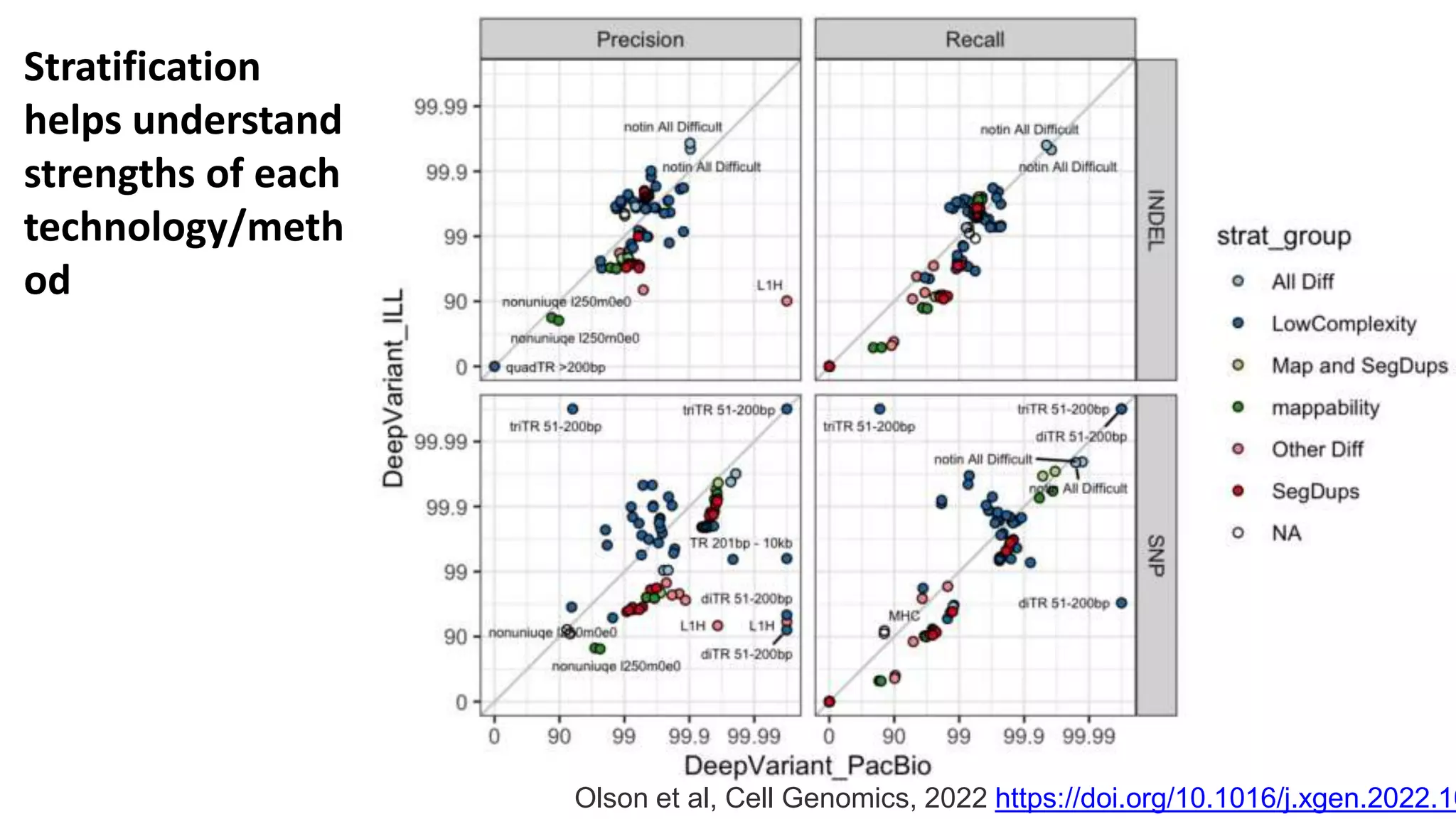
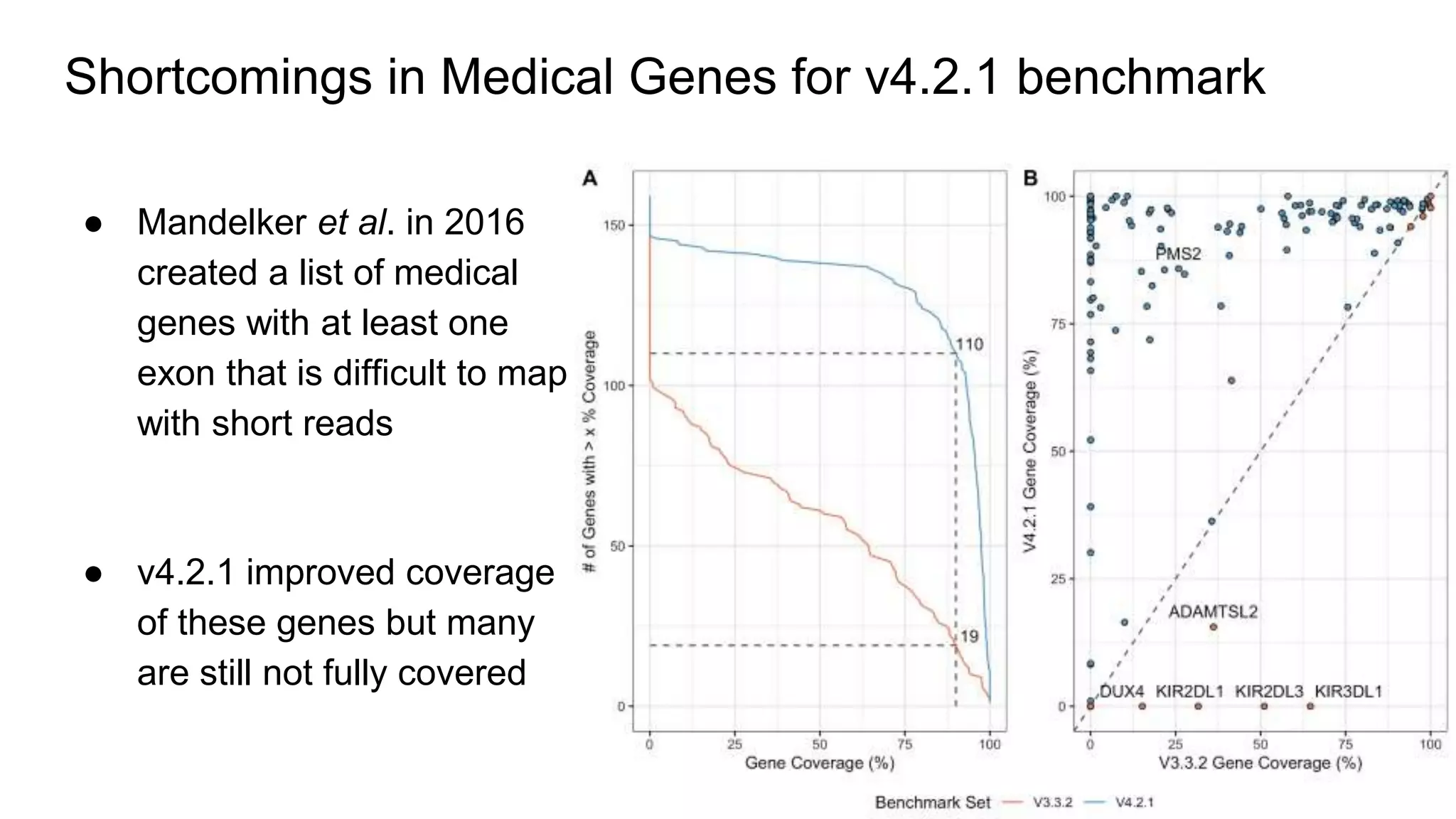
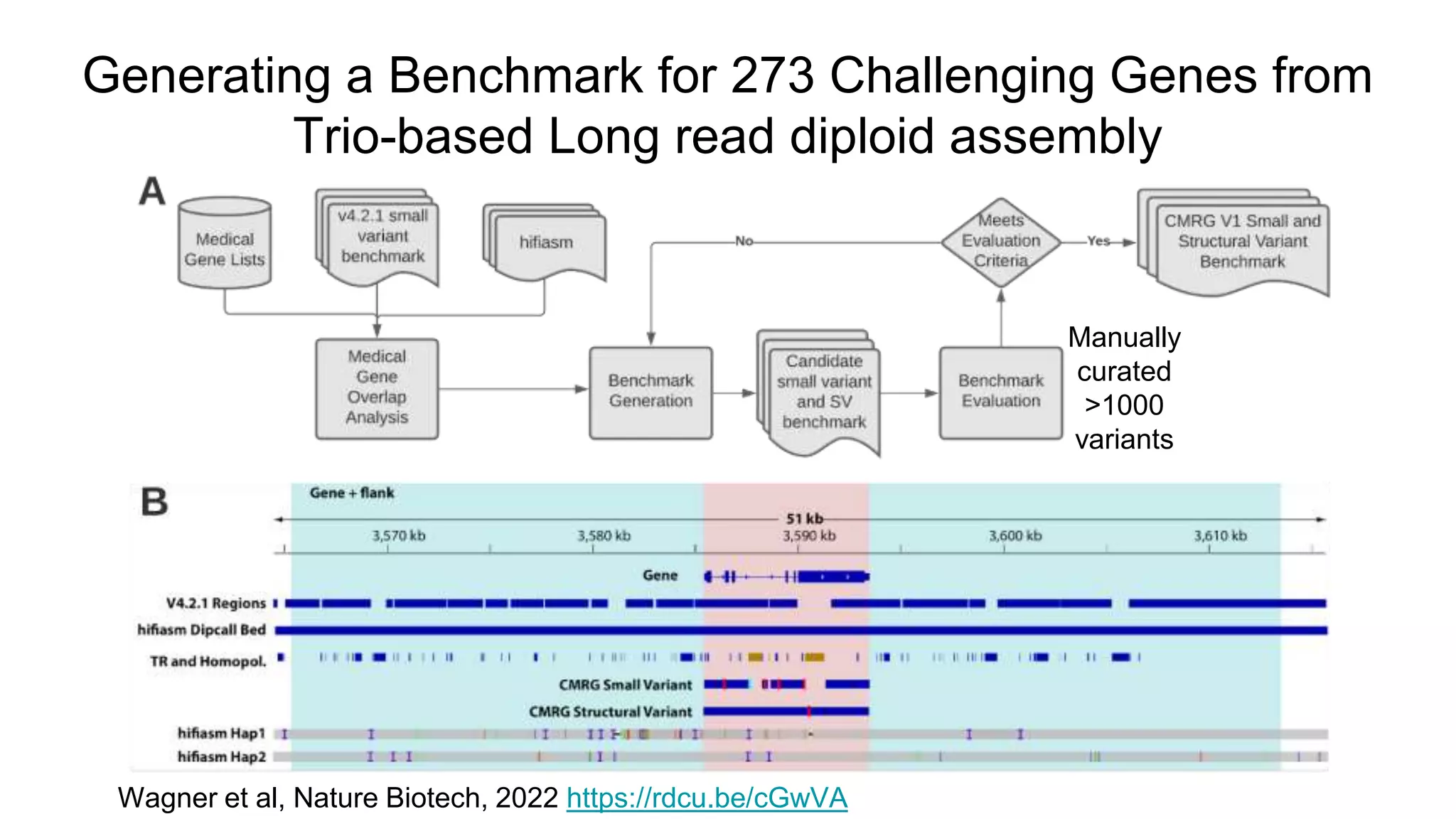
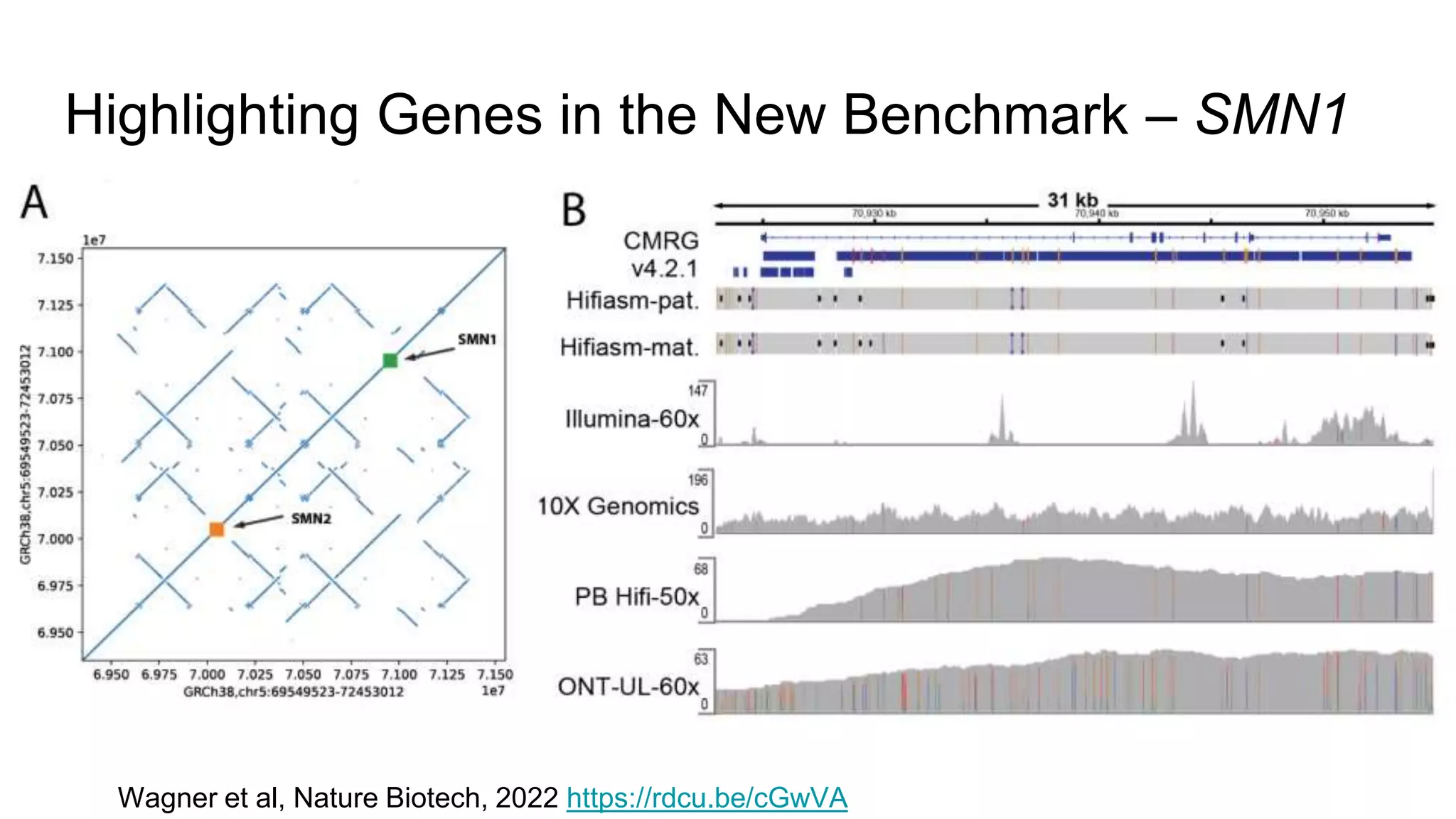

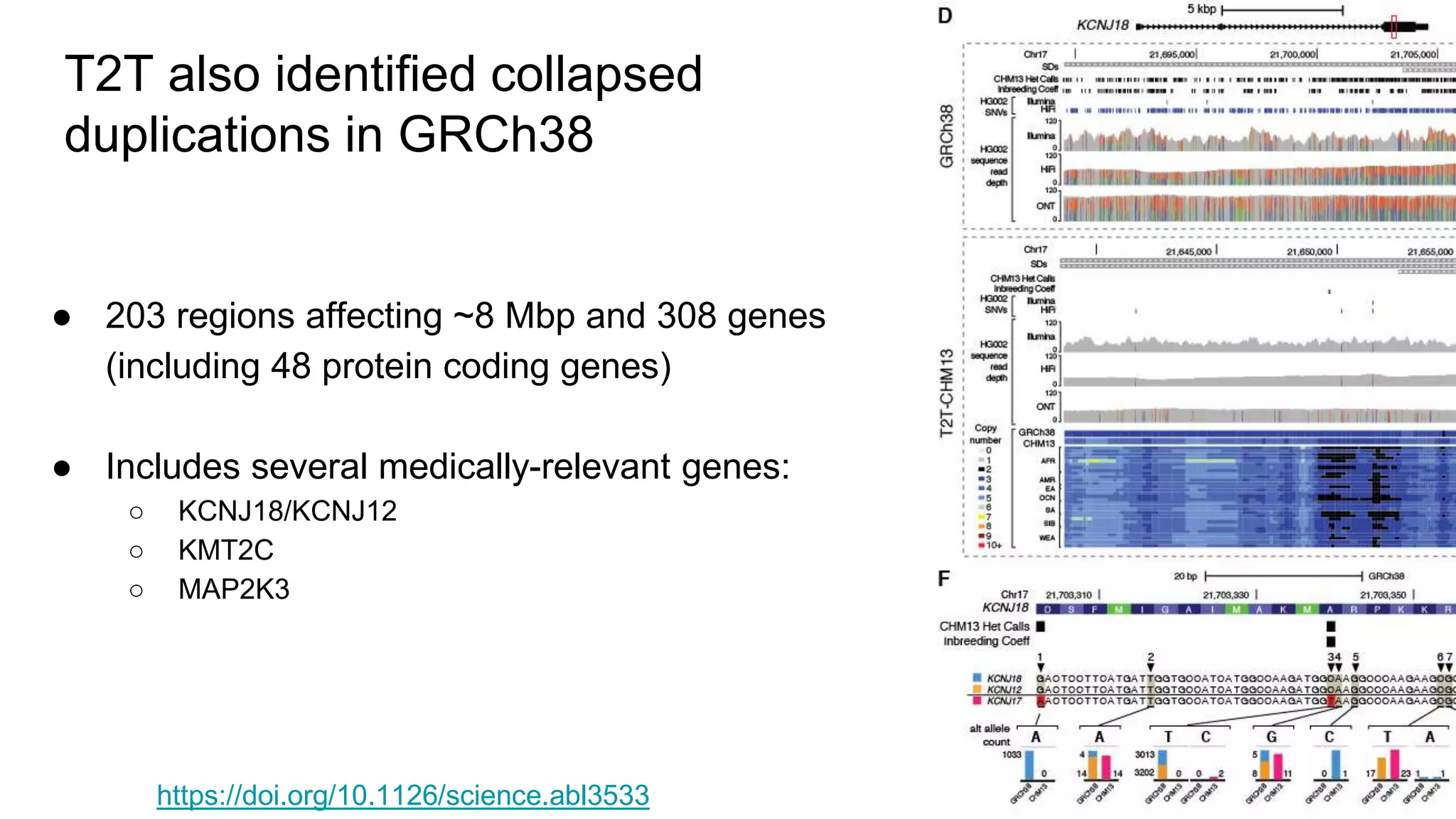



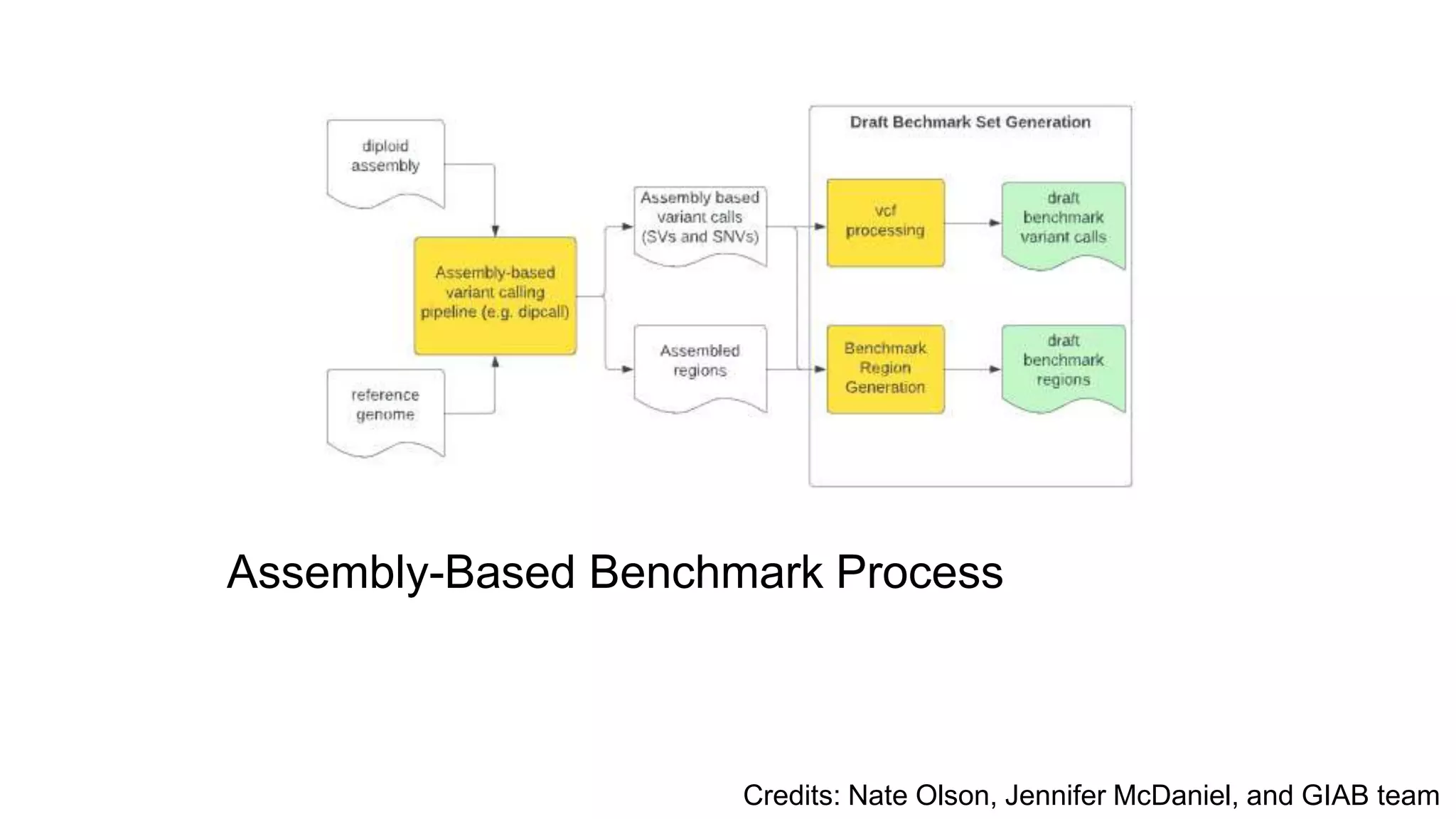





Using accurate long reads to improve Genome in a Bottle Benchmarks The Genome in a Bottle Consortium has used accurate long reads to characterize variants in difficult genomic regions for 7 human genomes. Long and linked reads improved the small variant benchmark by expanding reference coverage and the number of called variants. Accurate long reads were also essential for generating benchmarks for medically relevant genes and for improving benchmarks on chromosomes X and Y. Ongoing work includes developing RNA sequencing benchmarks from long reads and generating the first tumor/normal cell line benchmark.






















































































