Download to read offline



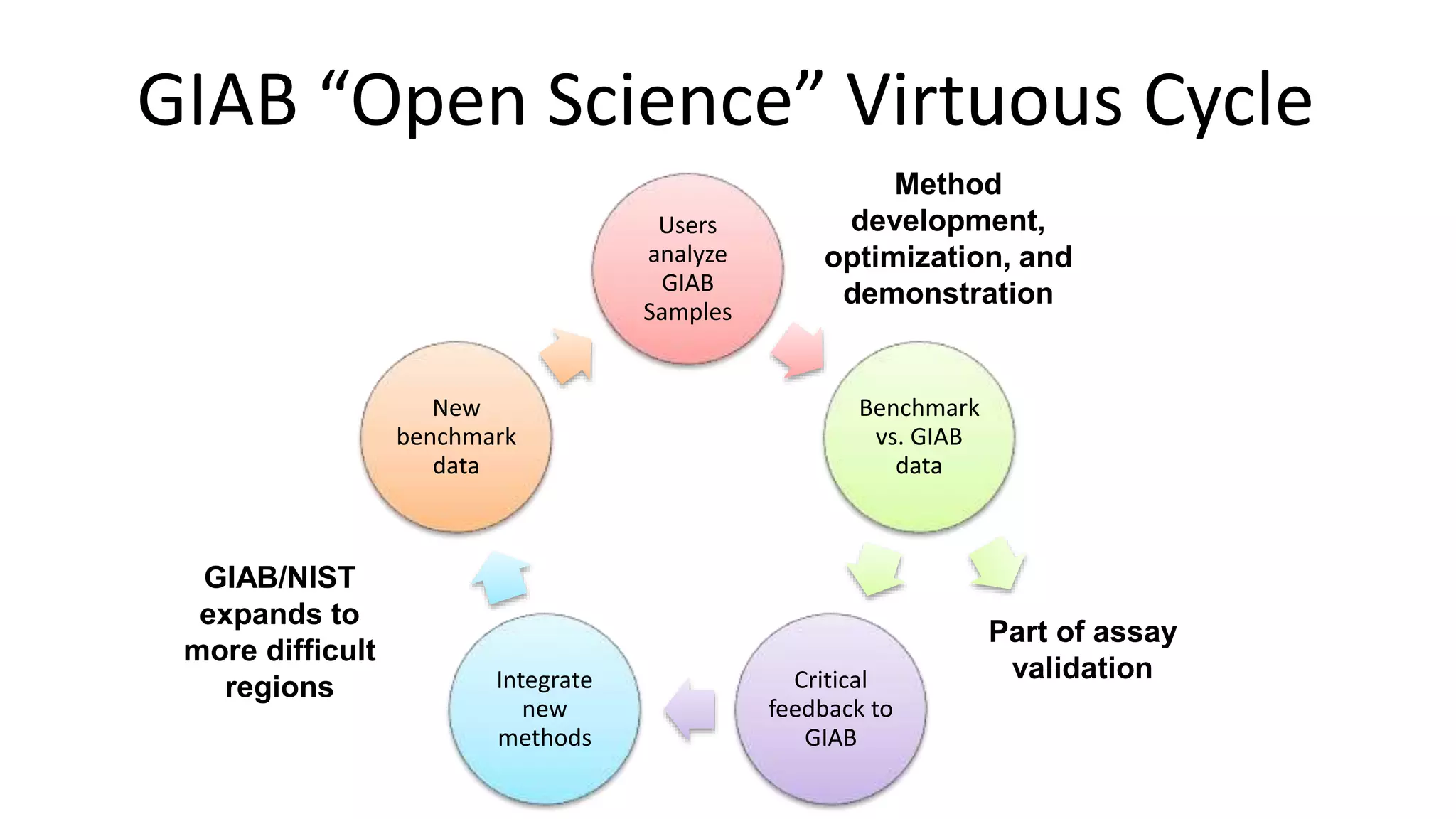
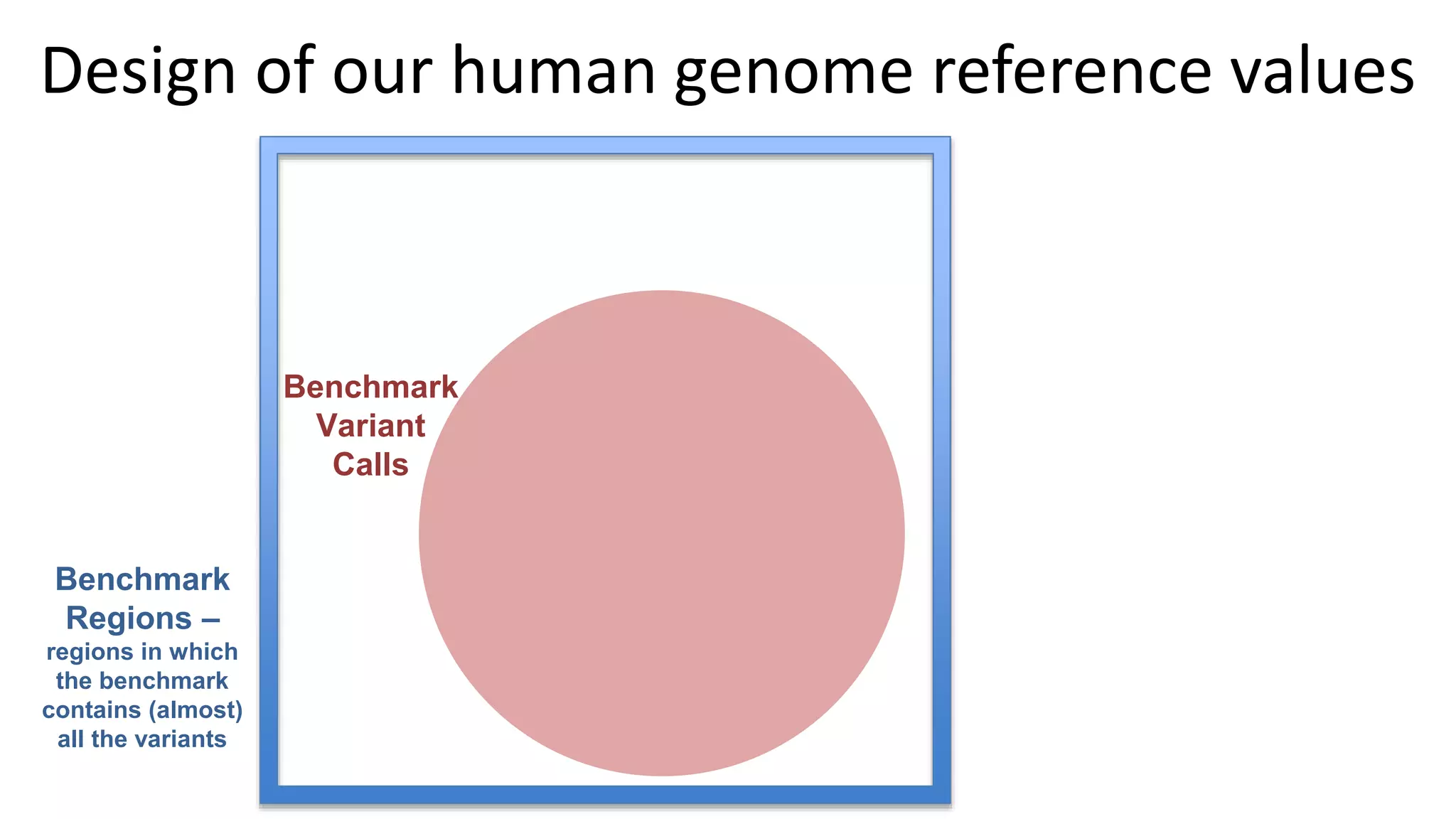
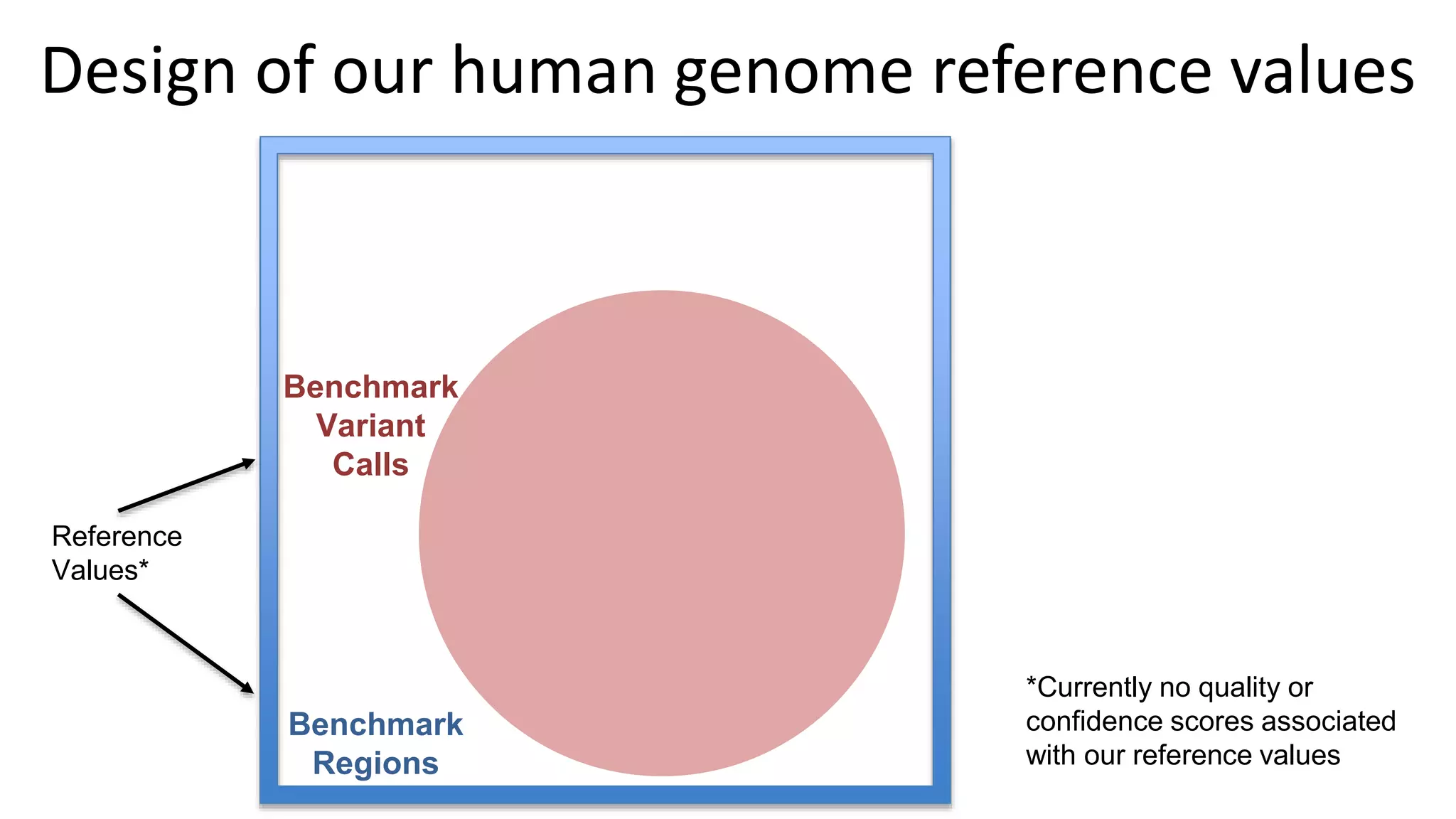
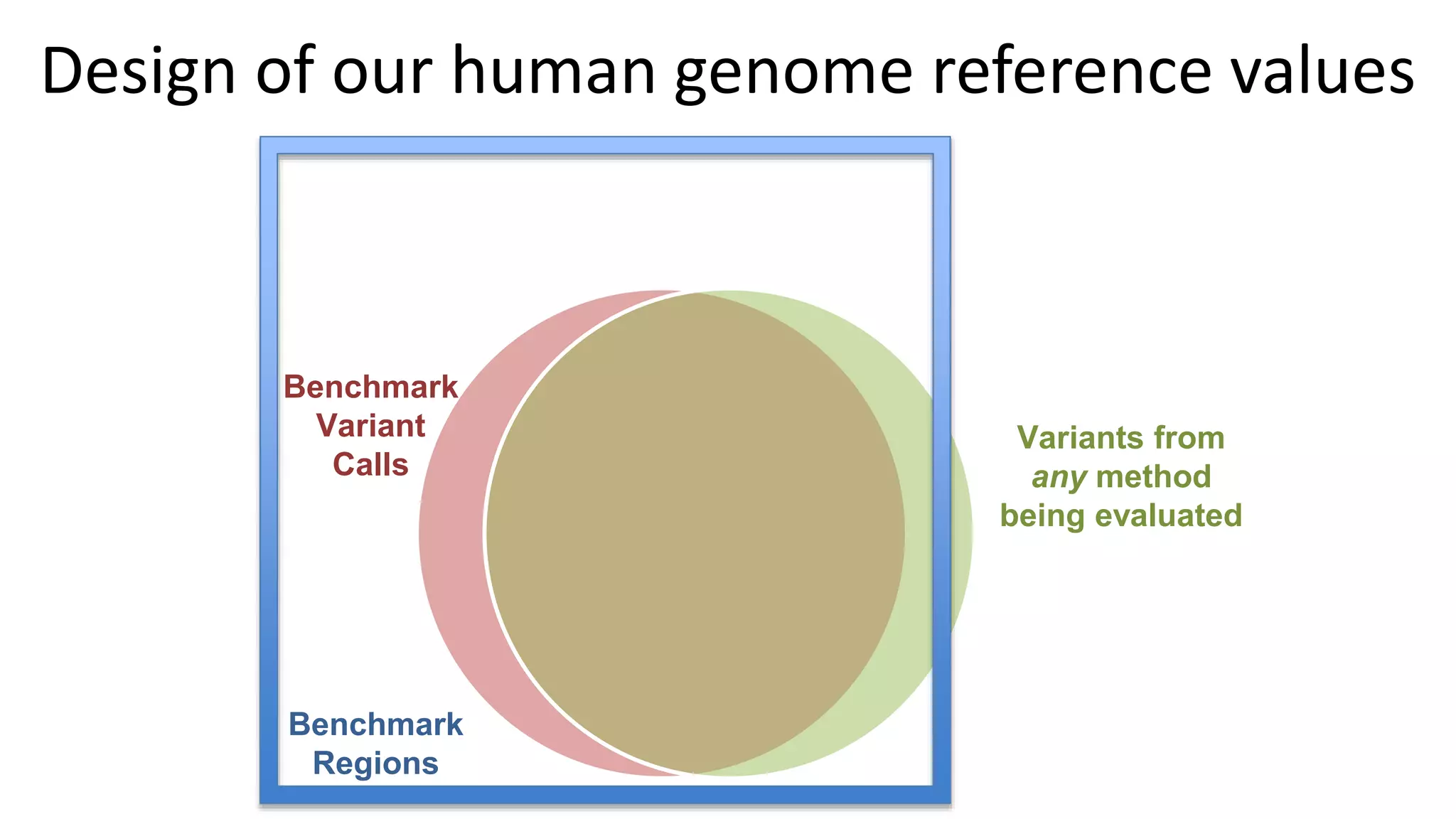
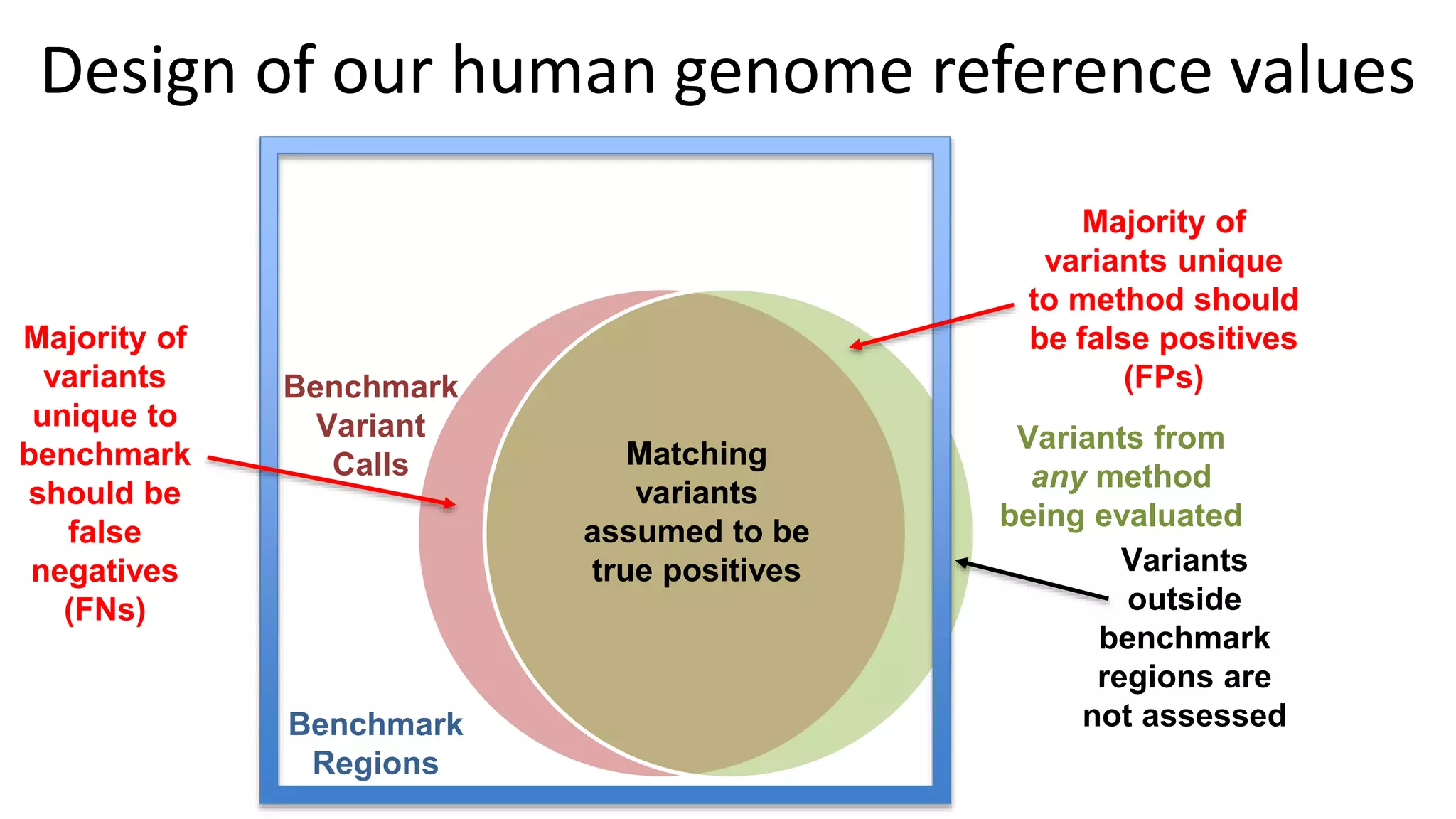



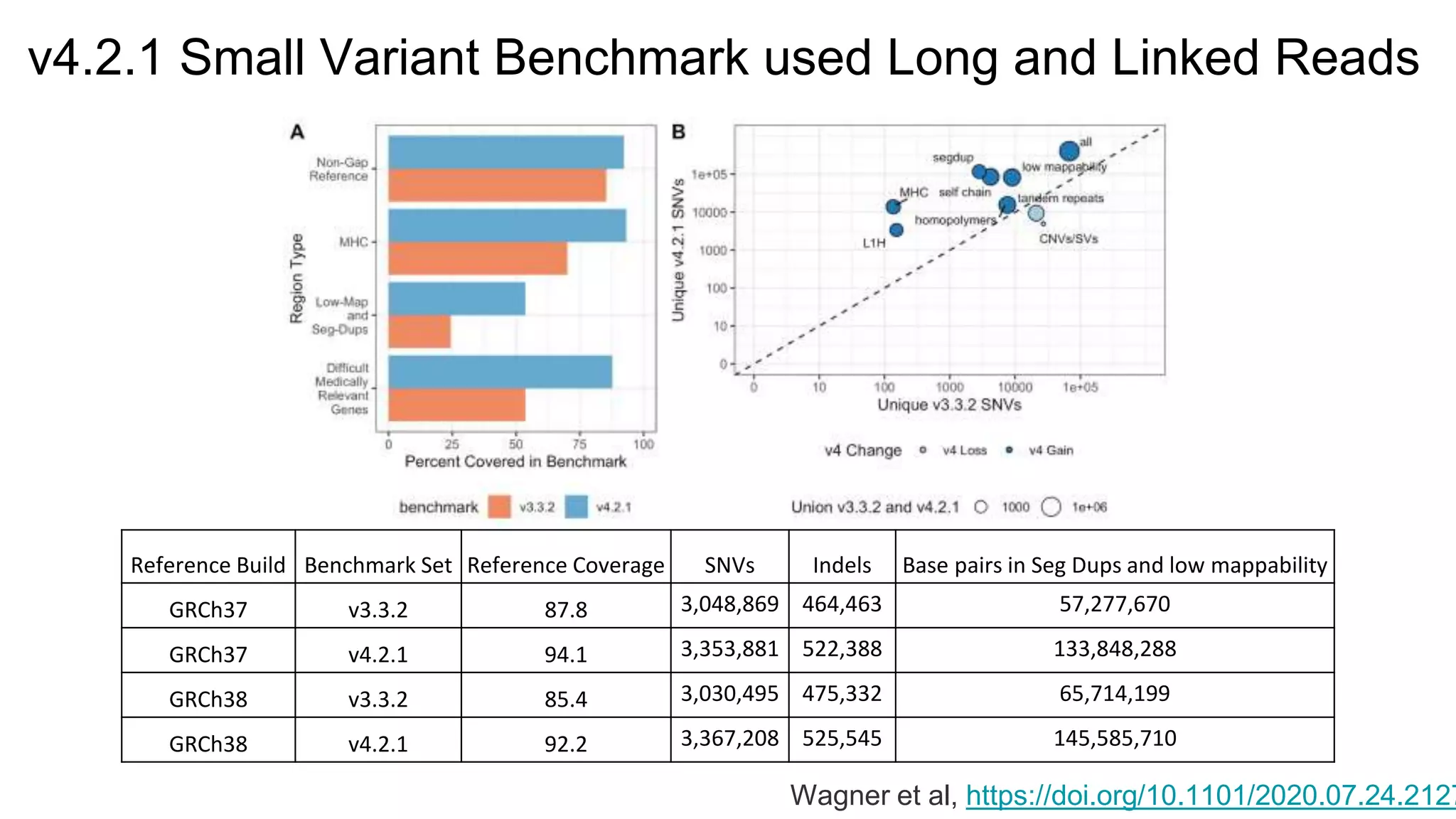
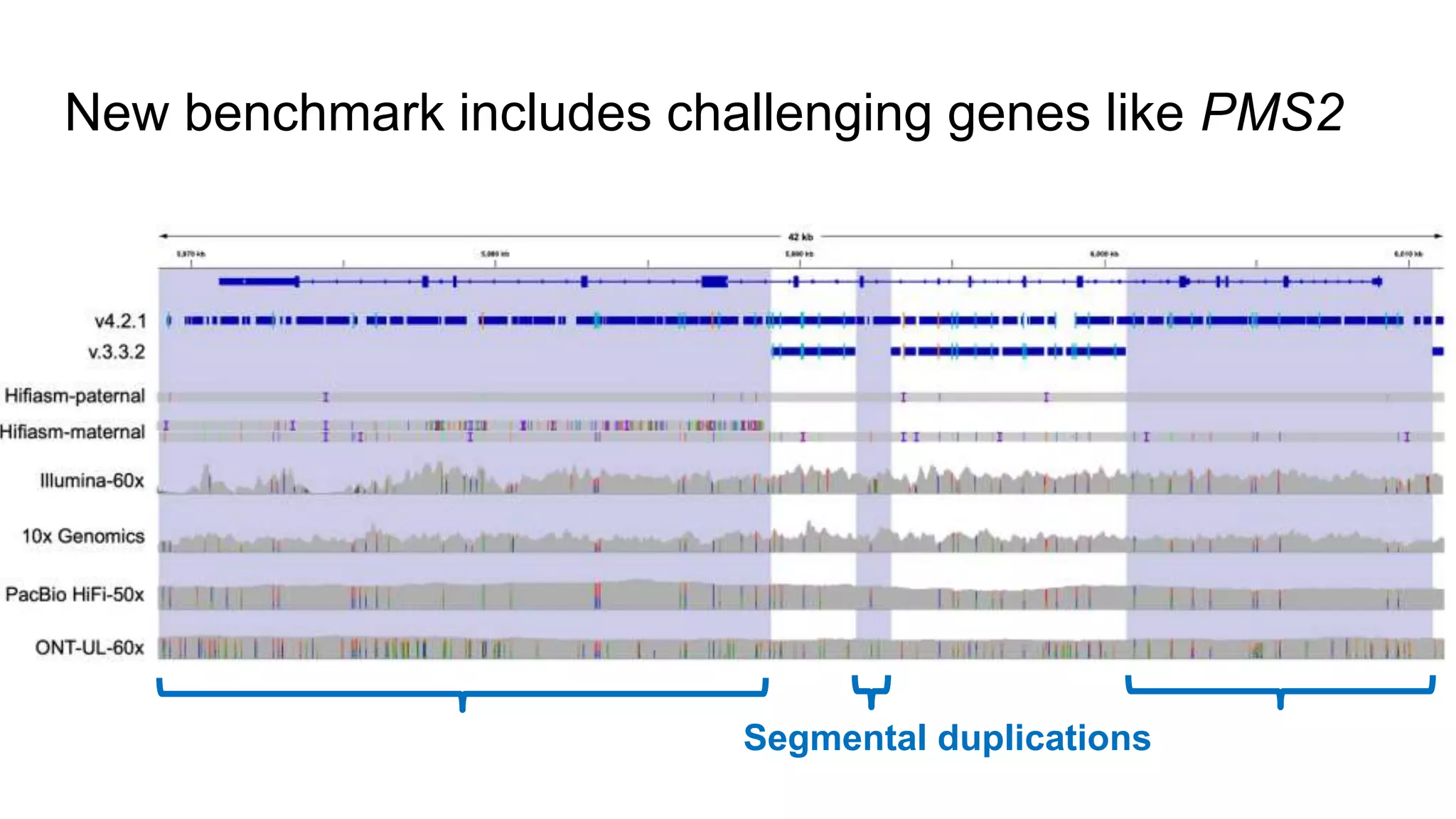


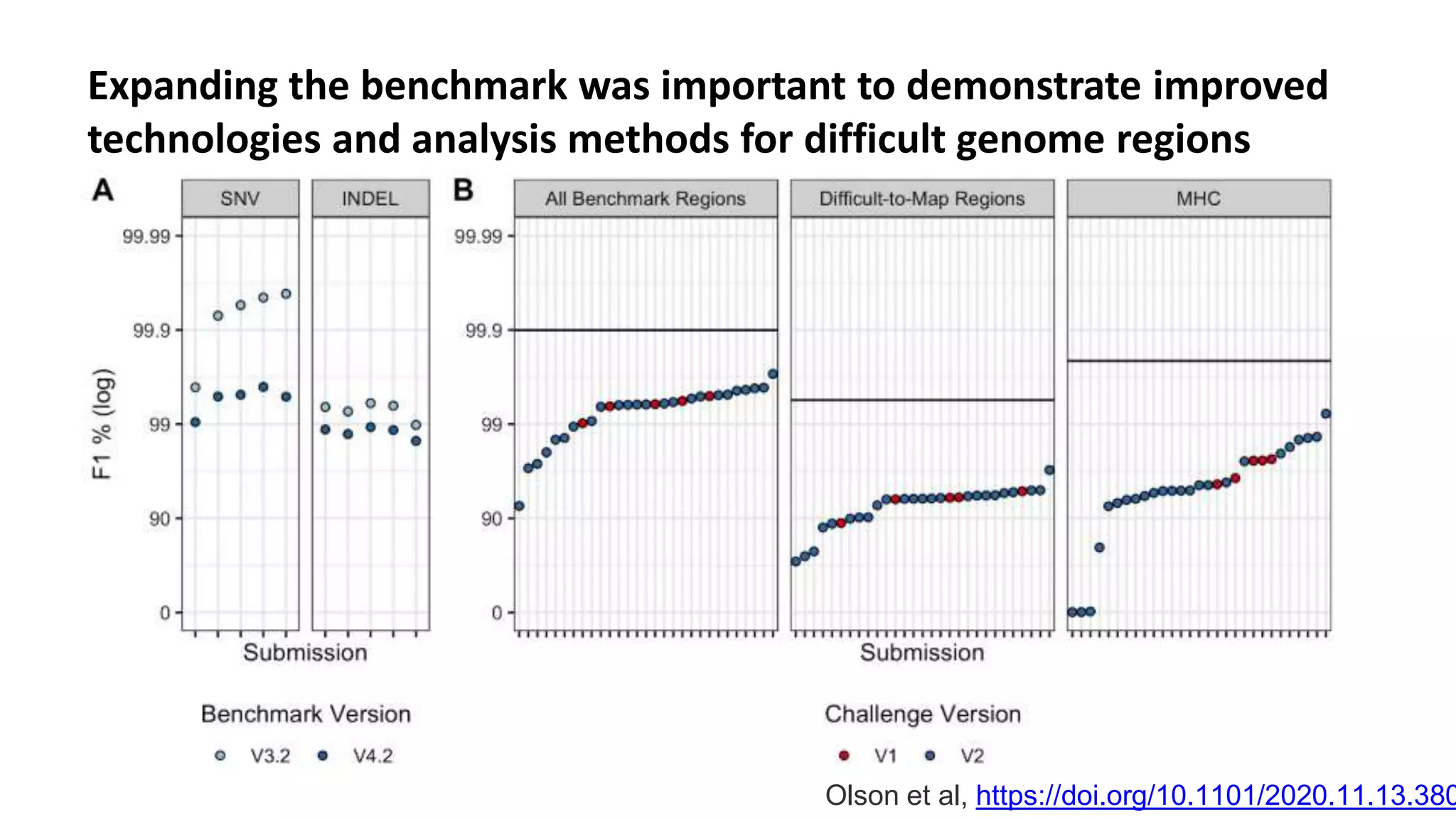

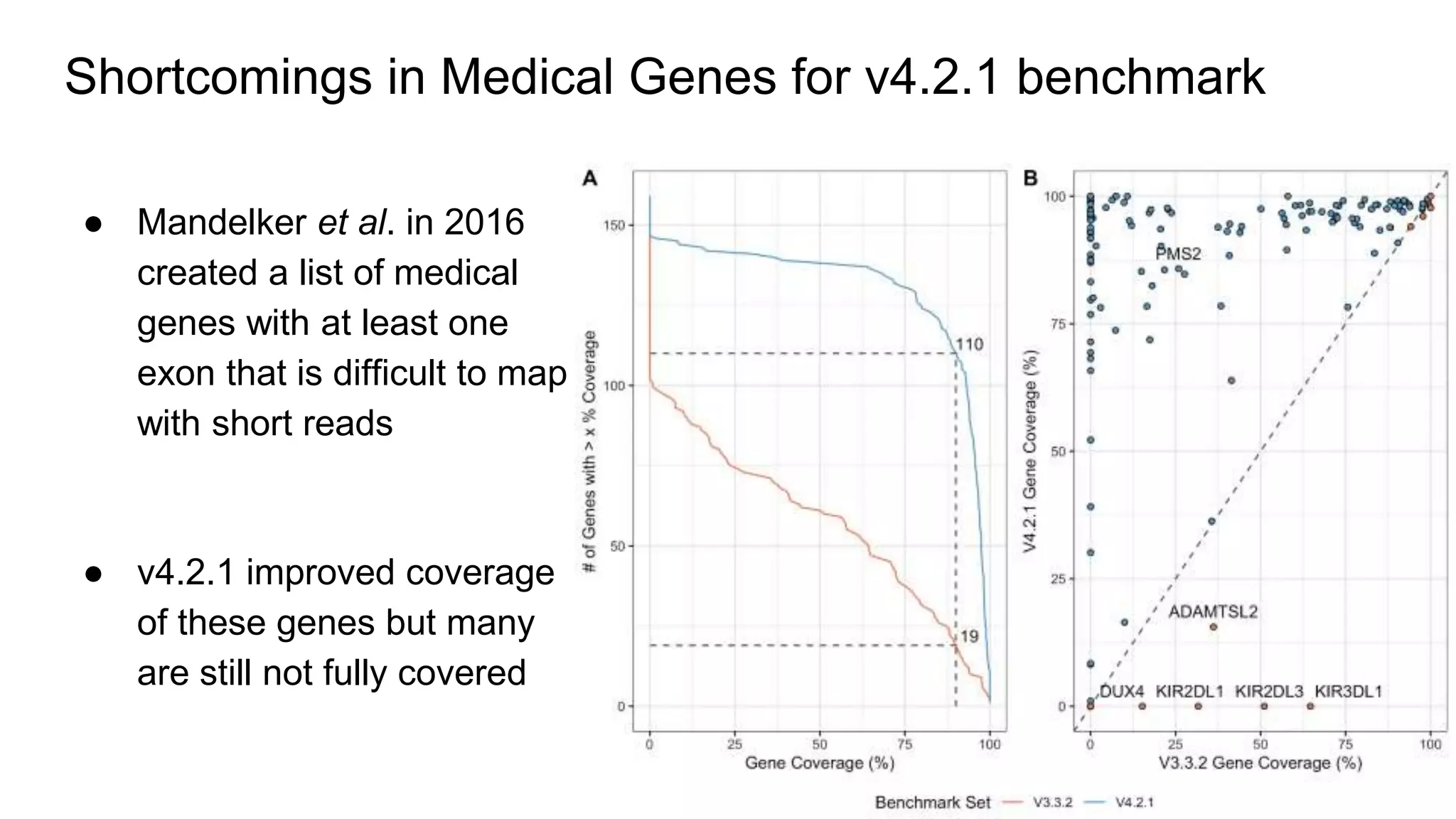

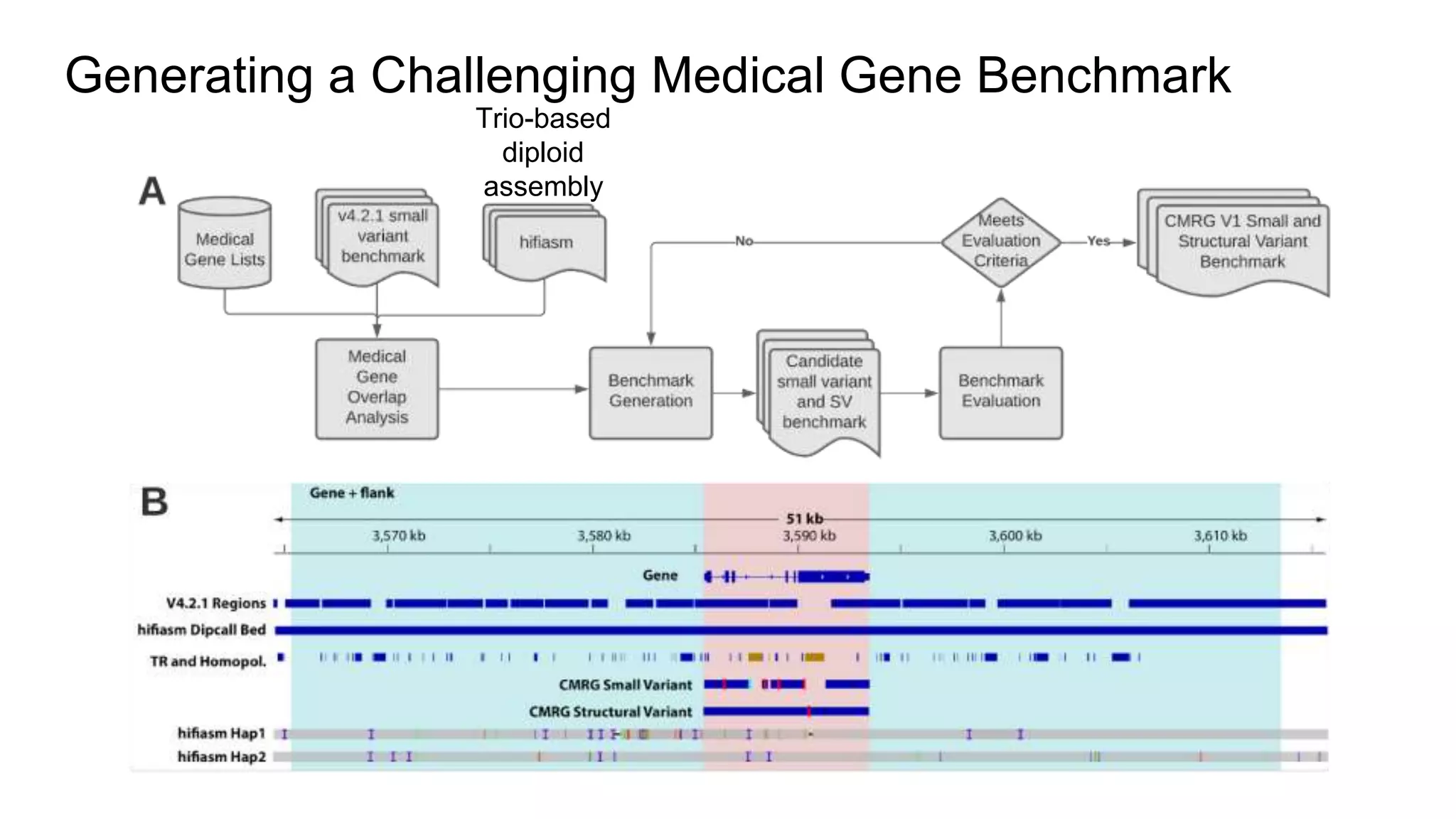
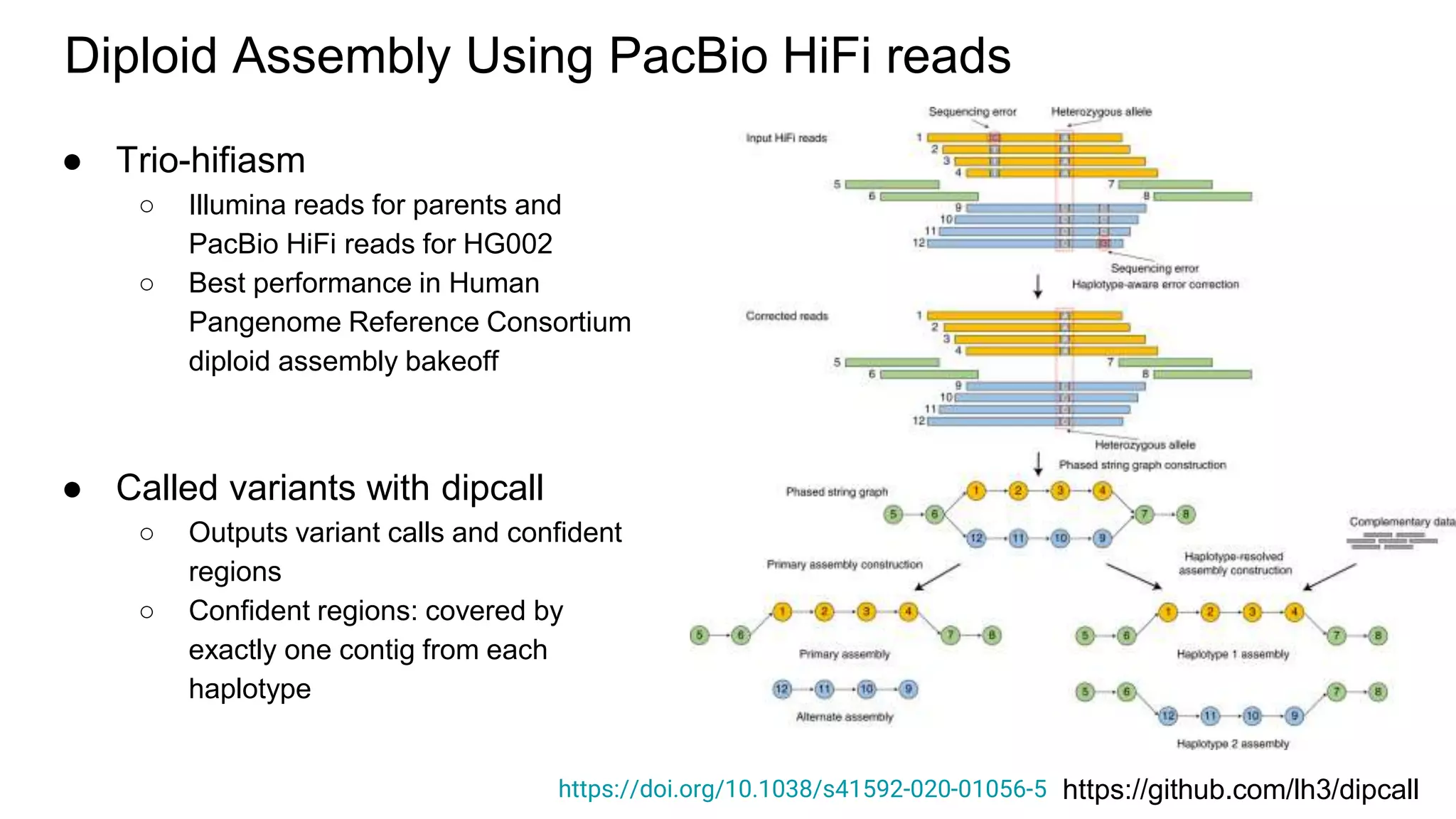
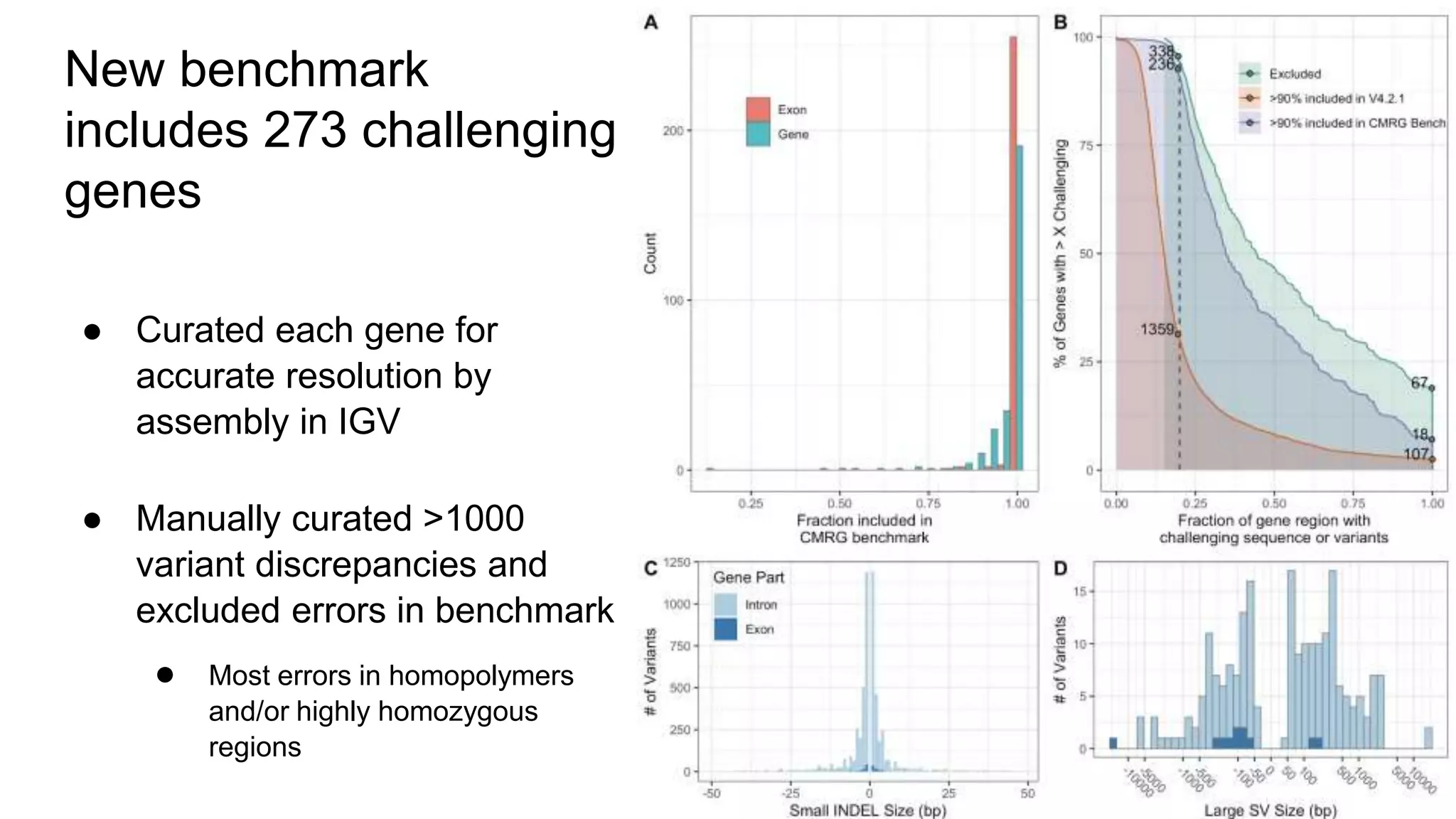

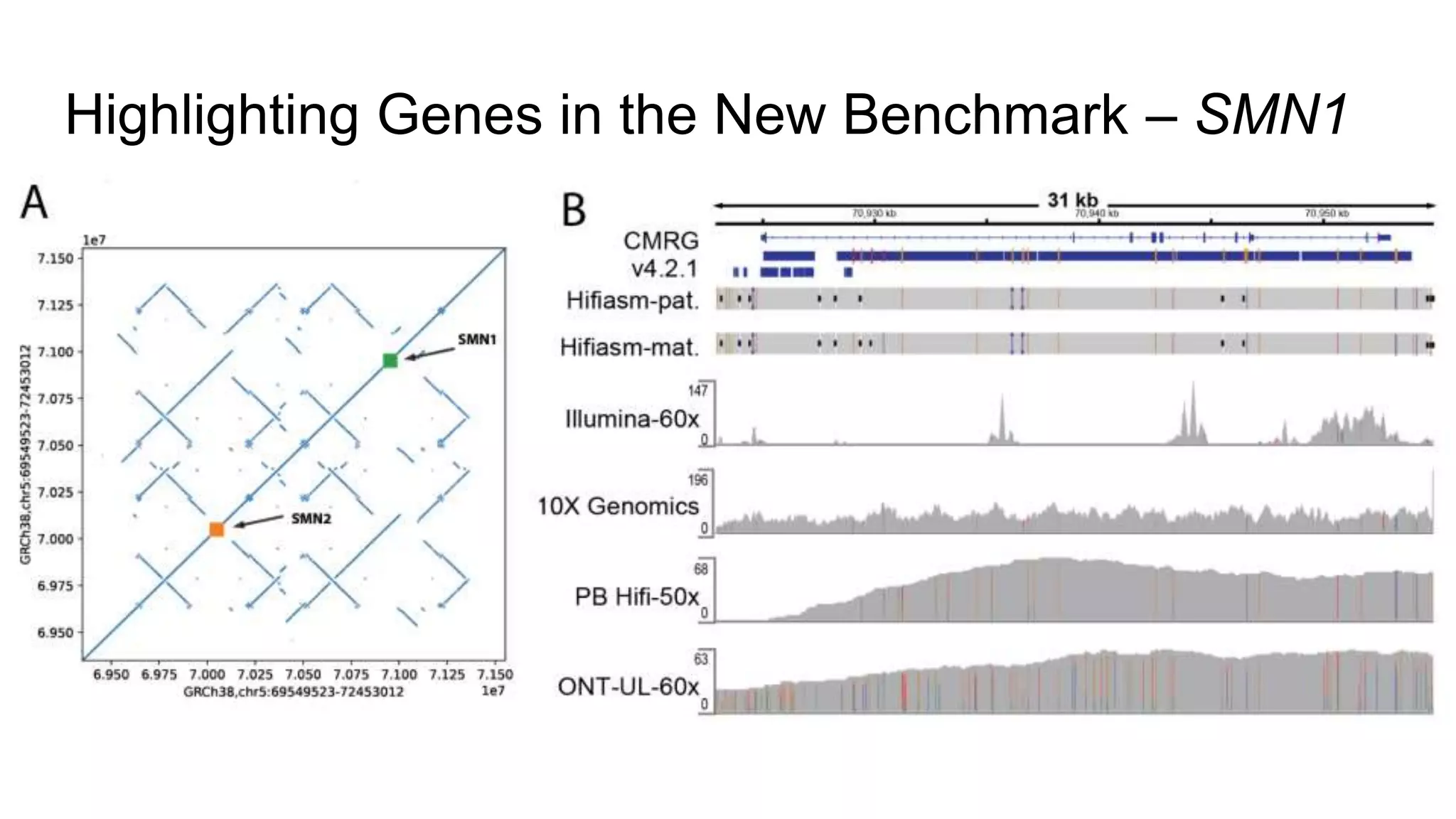




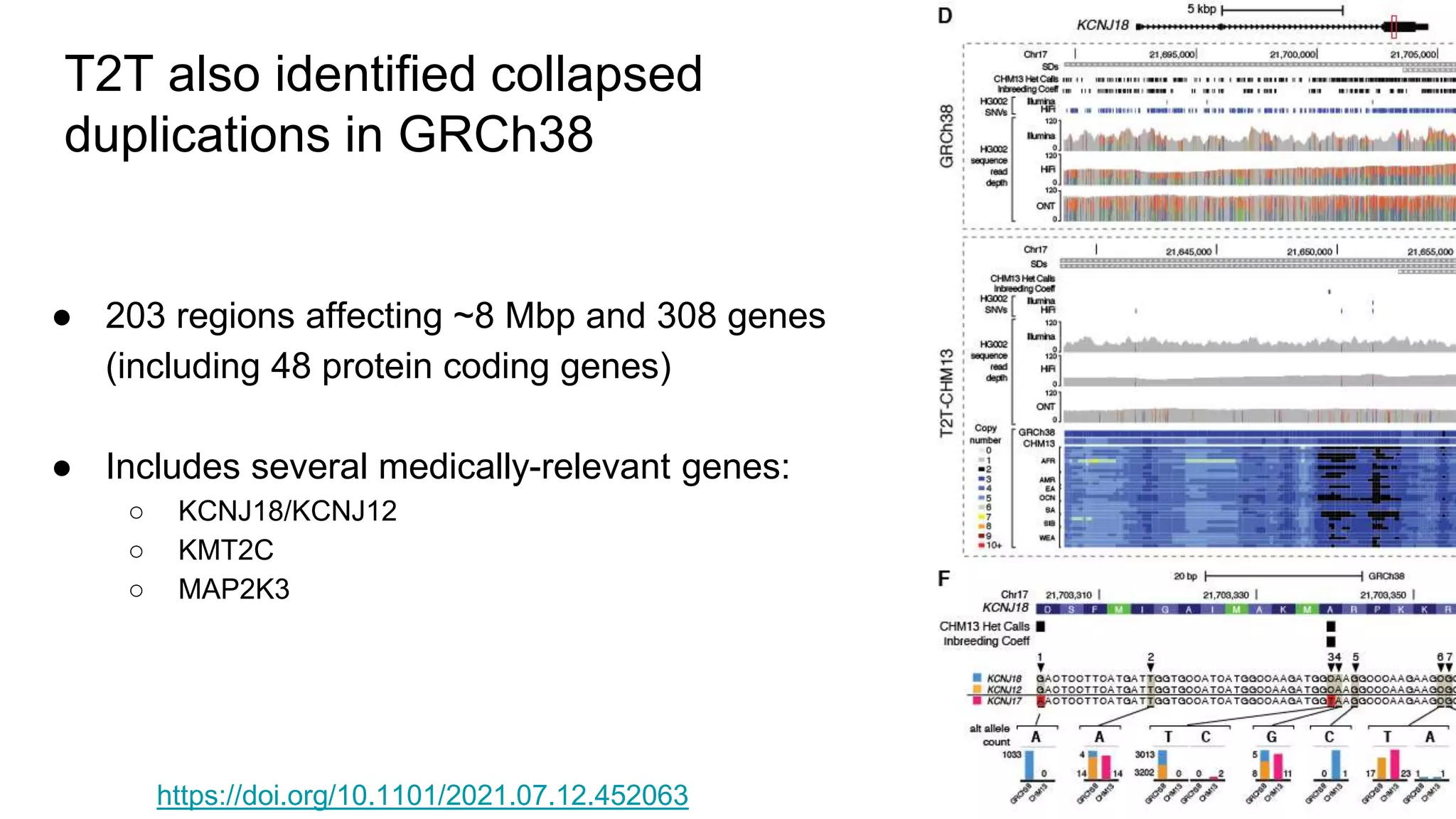


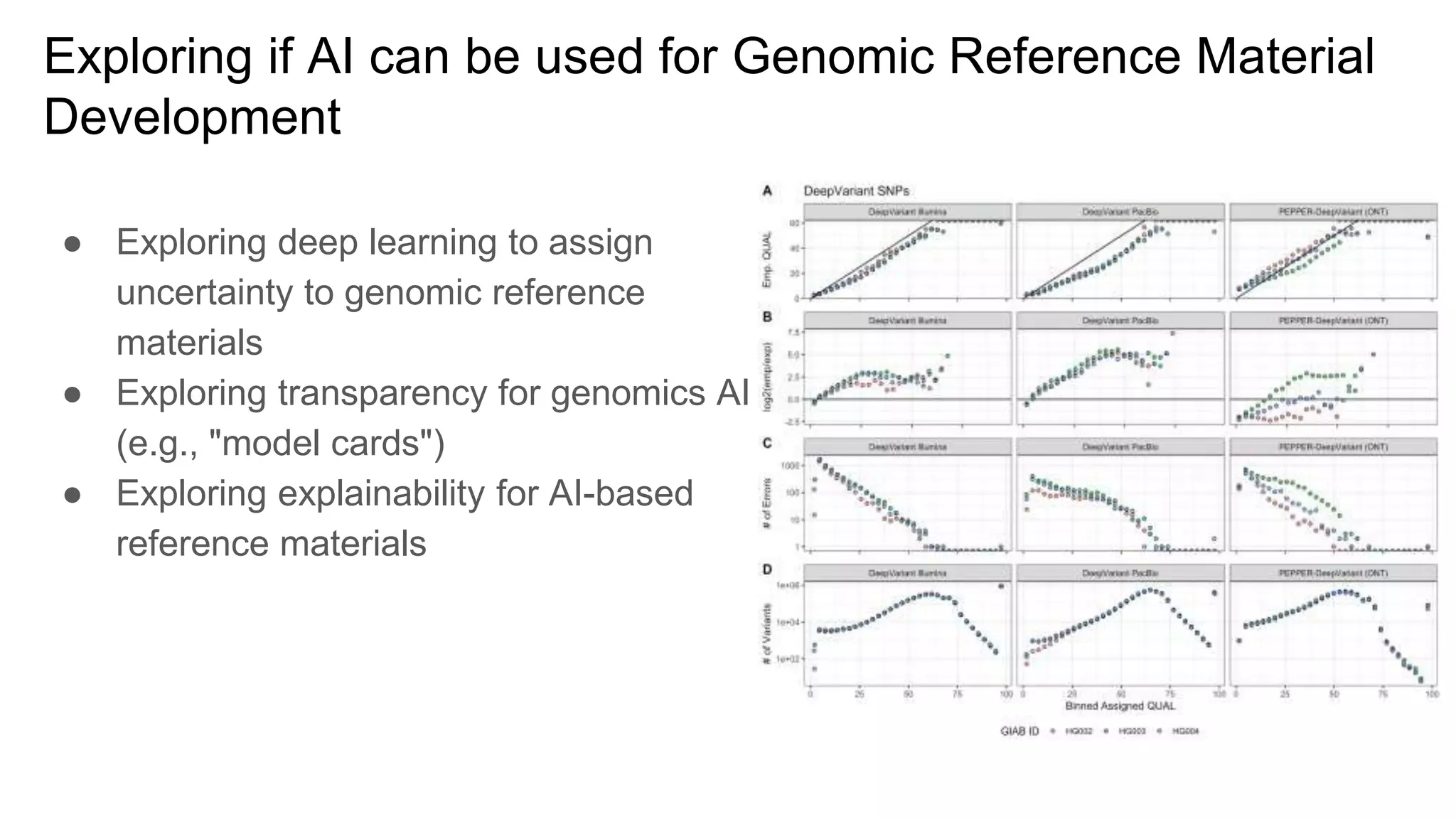




The 'Genome in a Bottle' initiative aims to establish reference materials for benchmarking human genome variant calling, specifically focusing on difficult genomic regions. It provides a reference material (RM 8391) consisting of human DNA from an individual of Eastern European Ashkenazi Jewish ancestry, intended for assessing sequencing performance across various applications. As part of ongoing improvements, new benchmarks are being developed to enhance the accuracy of variant assessments and tackle challenges in mapping complex genomic structures.






















































































