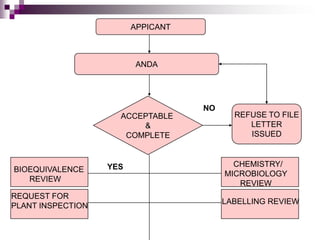
An Abbreviated New Drug Application (ANDA) contains data to allow the FDA to review and approve a generic drug. The ANDA establishes that the generic is bioequivalent to the reference drug through comparative dissolution or bioequivalence testing.
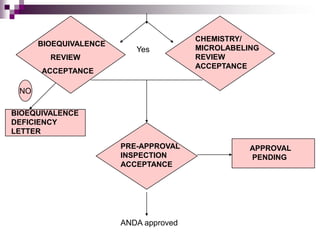
The FDA reviews the ANDA for bioequivalence, chemistry and manufacturing controls, microbiology, and labeling. If deficiencies are identified, the applicant is notified. If all aspects are acceptable, an approval or tentative approval is issued pending any patent issues. The generic can be marketed once all review steps are successfully completed.




















![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)


























![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)














































































