The document is a presentation on regulatory requirements for approval of biologics submitted by Arpitha B. M. to Dr. D. Manjula. It contains an introduction, history of biologics regulation citing key events, sources and types of biologics, differences between biologics and chemical drugs, the regulatory approval process including biological license application, and references. The presentation provides an overview of biologics, their regulation and approval process in India.


















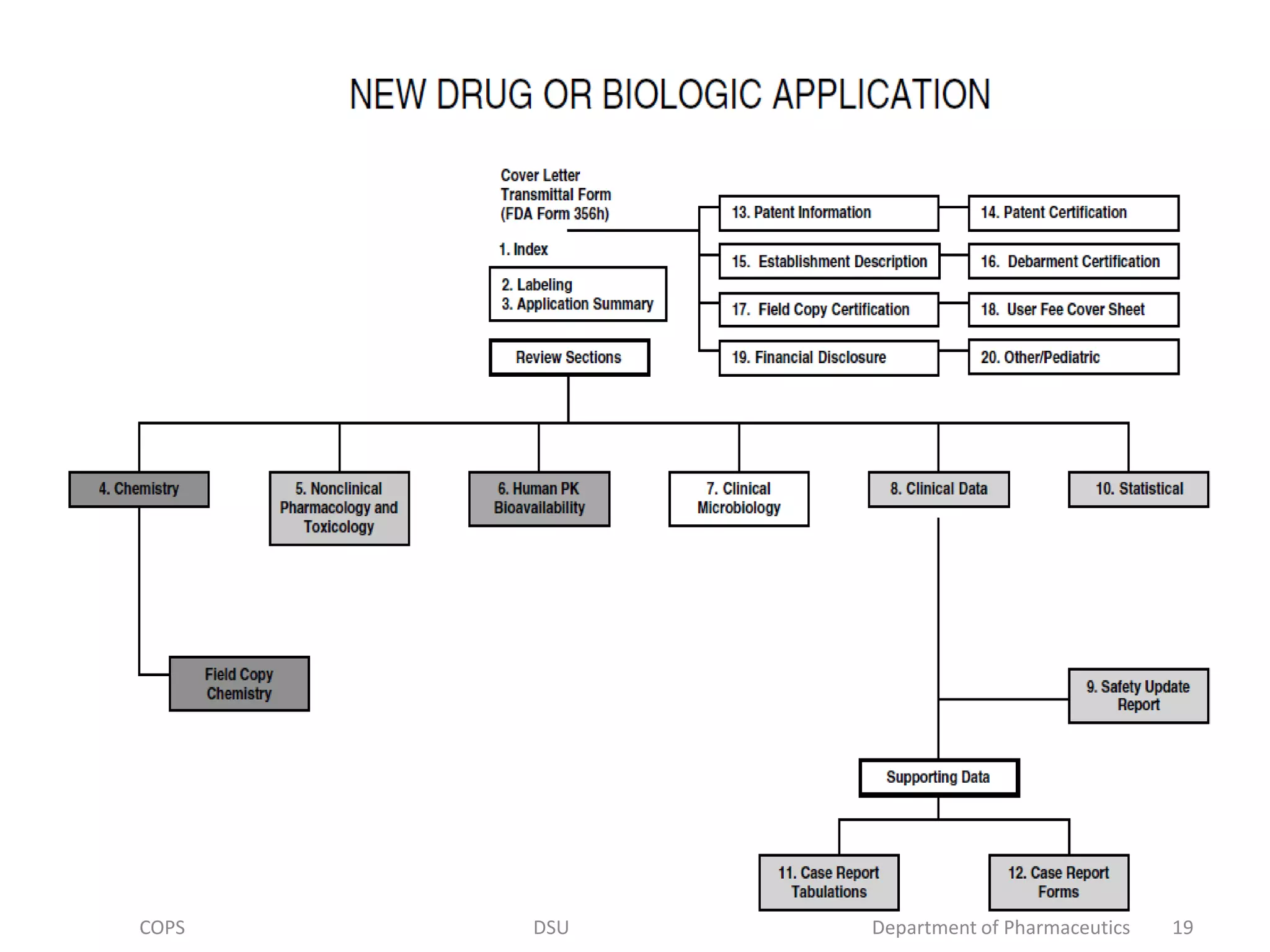
![• A cover letter should always accompany any FDA submission.
Addressed below are the Form FDA 356(h), the cover letter, and all
20 sections of the BLA application.
• Before each section is addressed individually, it is worth
emphasizing the importance of the application form [Form FDA
356(h)], the cover letter, and the first three sections.
• Cover Letter: It consists of basic administrative information
requested about the BLA application (e.g., sponsor name and
address, etc.). The cover letter should provide at least seven types of
information:
1. Name and address of sponsor and others
2. Product name
3.Reason for submission(e.g original submission, supplement,
amendment, etc.).
4.Information contained in the submission..
5.Agreements with the FDA.
6.Other documents relating to submission.
7.special circumstances.
8.Fast track review.
COPS DSU Department of Pharmaceutics 20](https://image.slidesharecdn.com/biologics-191119133905/75/Regulatory-requirement-for-approval-of-Biologics-20-2048.jpg)












![REVISED Guidance for Industry: Providing
Regulatory Submissions to the Center for Biologics
Evaluation and Research (CBER) in Electronic Format-
Biologics Marketing Applications [Biologics License
Application (BLA), Product License Application (PLA)/
Establishment License Application (ELA) and New
Drug Application (NDA)] — November 12, 1999.
• Draft Guidance for Industry: Submitting Marketing
Applications According to the ICH-CTD Forma
General Considerations - September 5, 2001
COPS DSU Department of Pharmaceutics 33](https://image.slidesharecdn.com/biologics-191119133905/75/Regulatory-requirement-for-approval-of-Biologics-33-2048.jpg)





















































































