Downloaded 19 times





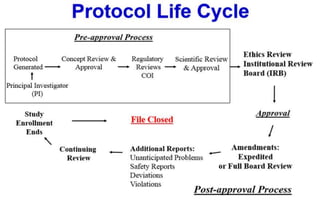






























This document outlines the key elements of a clinical trial protocol. It discusses that a protocol establishes the guidelines for conducting a clinical trial according to a pre-specified plan. The protocol describes all aspects of the trial, including objectives, design, methodology, statistical considerations and organization. It provides a systematic written plan to ensure the scientific integrity of the trial and protect the safety and rights of human subjects. The protocol contains administrative information, background, objectives, methodology, safety monitoring and ethical considerations.























![INVESTIGATIONAL NEW DRUG [IND] APPLICATION SUBMISSION (1).pdf](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugindapplicationsubmission1-221128141339-adad00bd-thumbnail.jpg?width=640&height=640&fit=bounds)






![Indian gcp guidelines[647]](https://cdn.slidesharecdn.com/ss_thumbnails/indiangcpguidelines647-210325044800-thumbnail.jpg?width=640&height=640&fit=bounds)

















































