Downloaded 14 times



























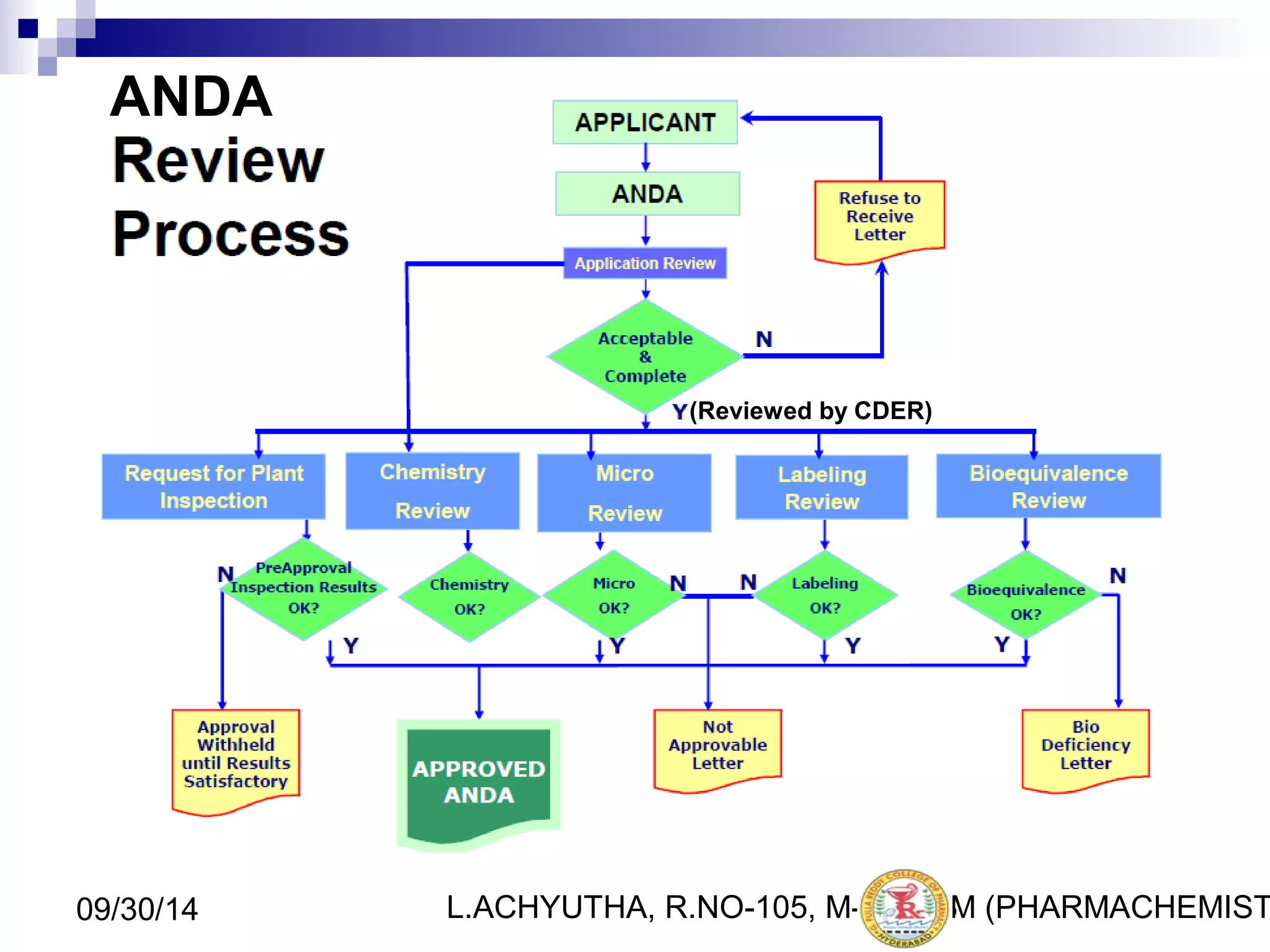









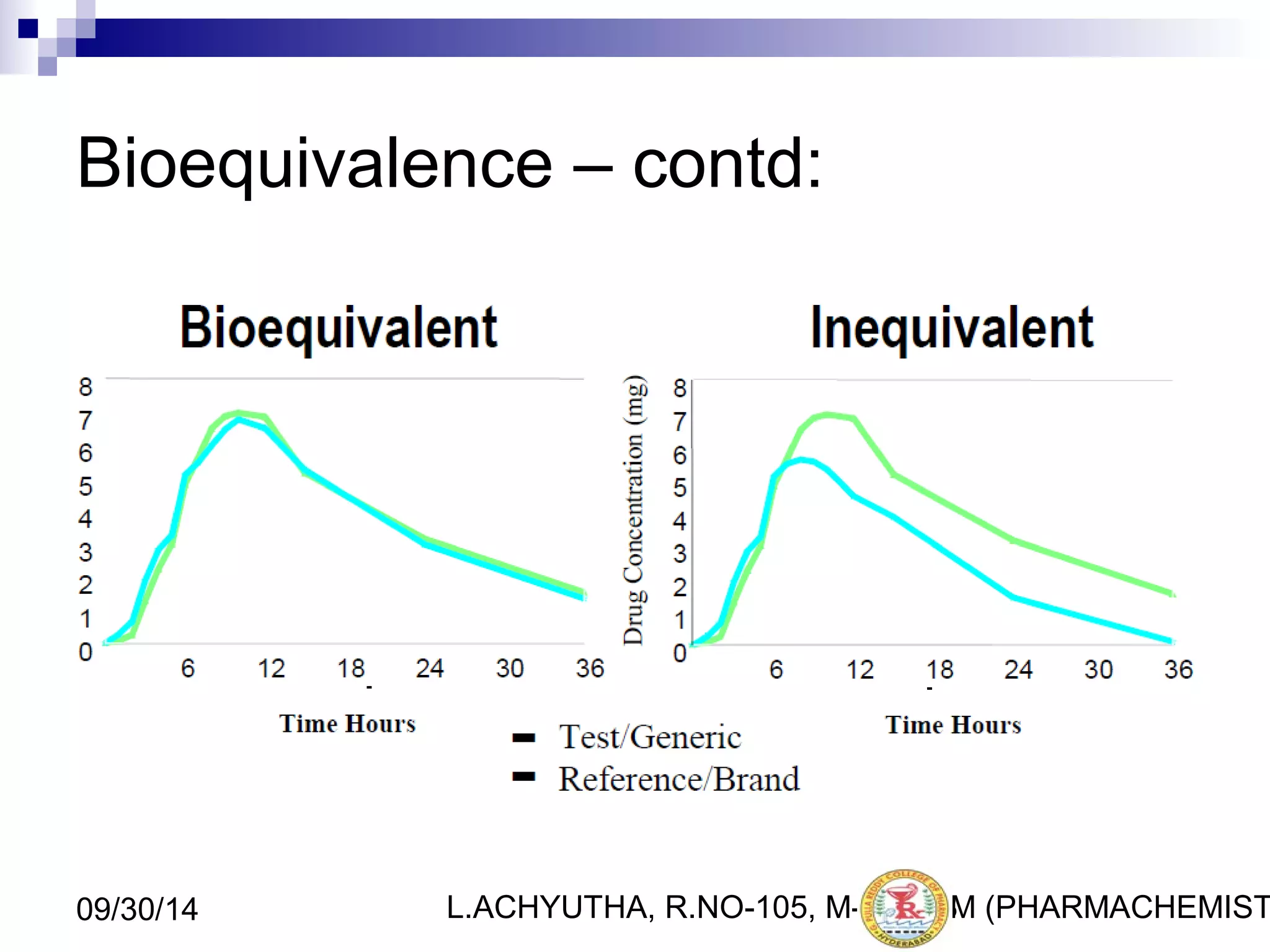
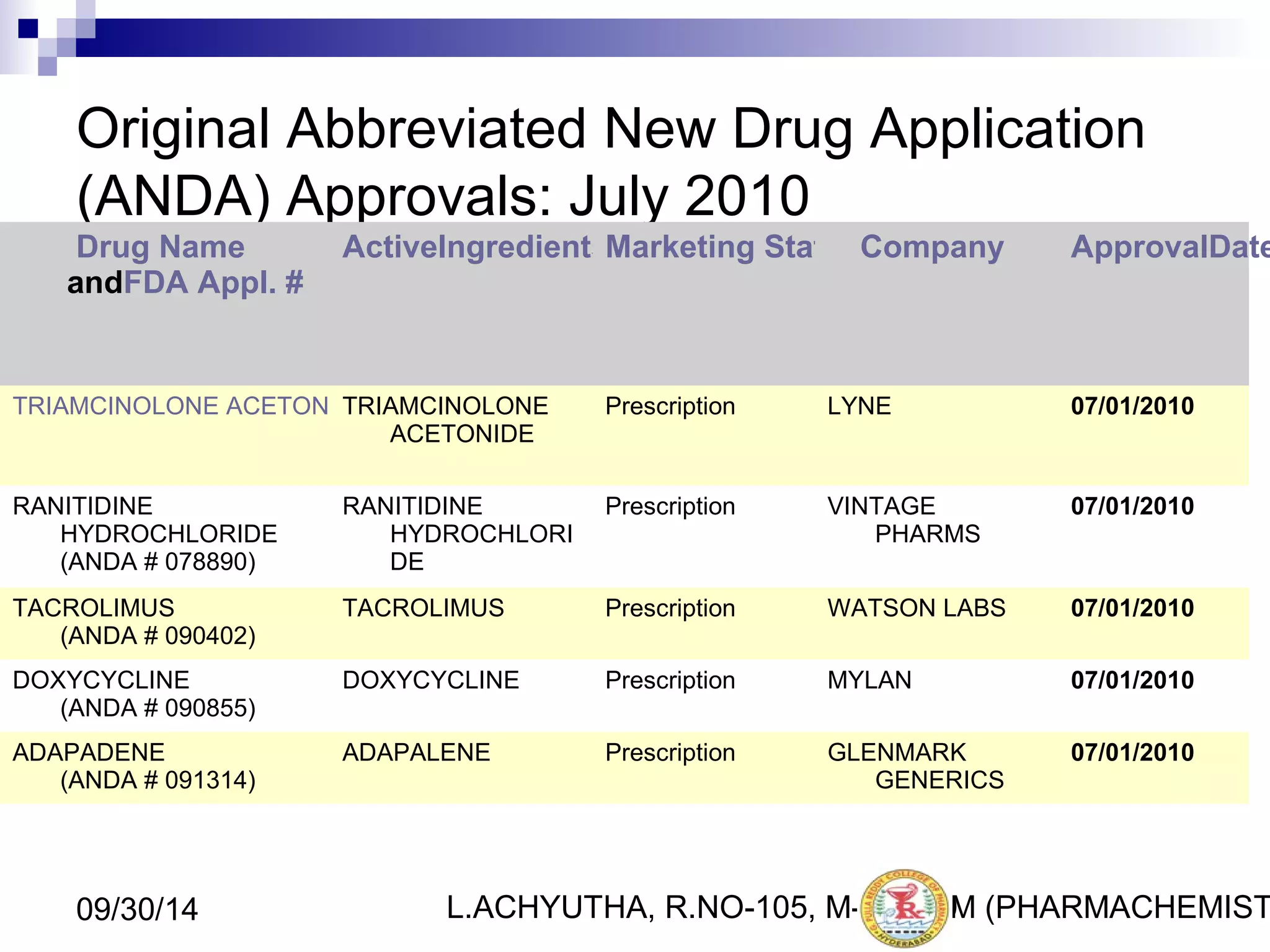




An ANDA (Abbreviated New Drug Application) is an application for approval of a generic drug that is comparable to an existing approved brand-name drug in dosage form, strength, route of administration, quality and performance characteristics. An ANDA contains data to demonstrate that the generic drug is bioequivalent to the reference listed drug but does not require clinical data of safety and efficacy. The ANDA review process by the FDA's Center for Drug Evaluation and Research is abbreviated compared to a New Drug Application as it focuses on demonstrating bioequivalence rather than independent evidence of safety and efficacy.































![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)



































