Download as PDF, PPTX





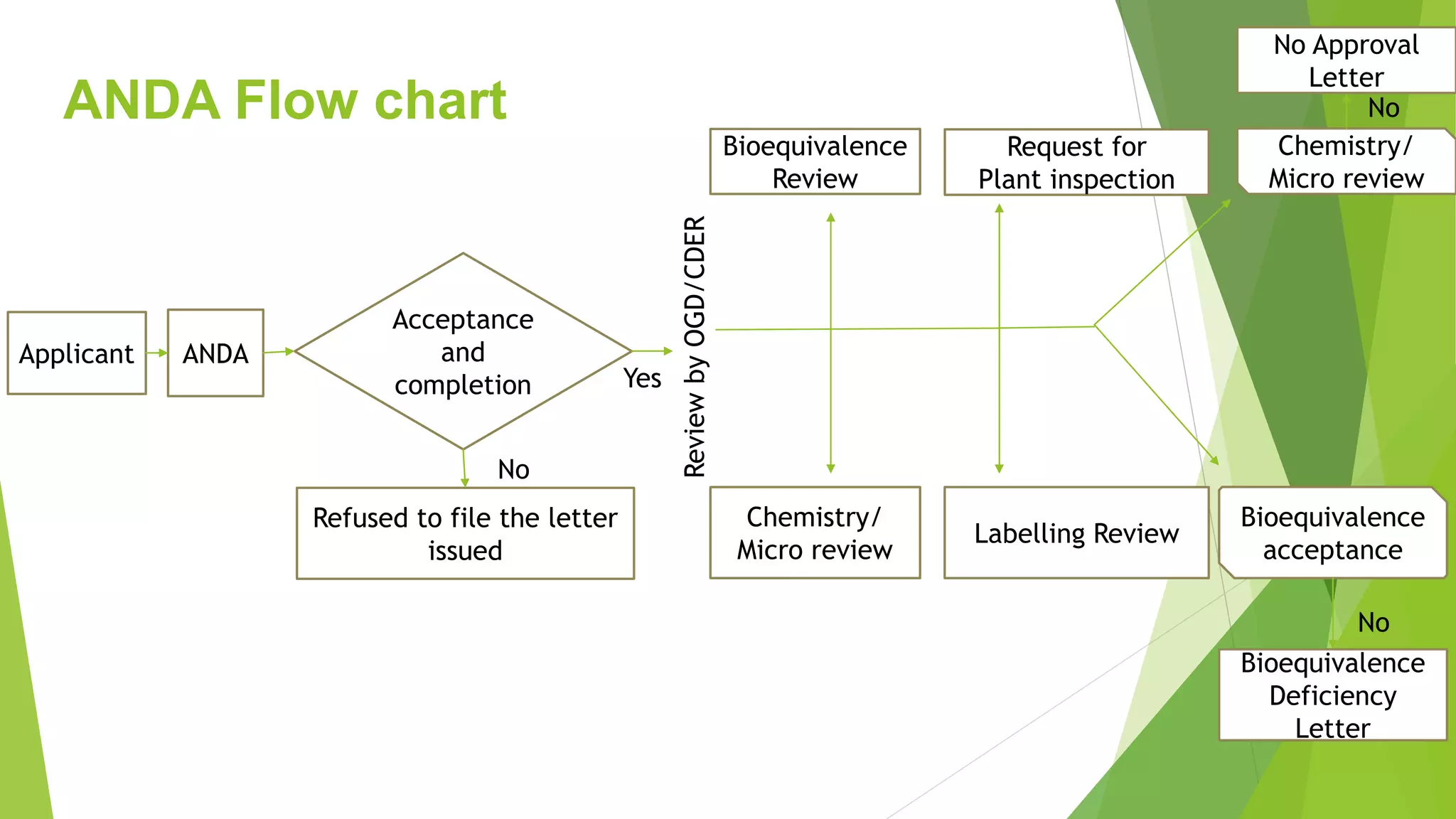
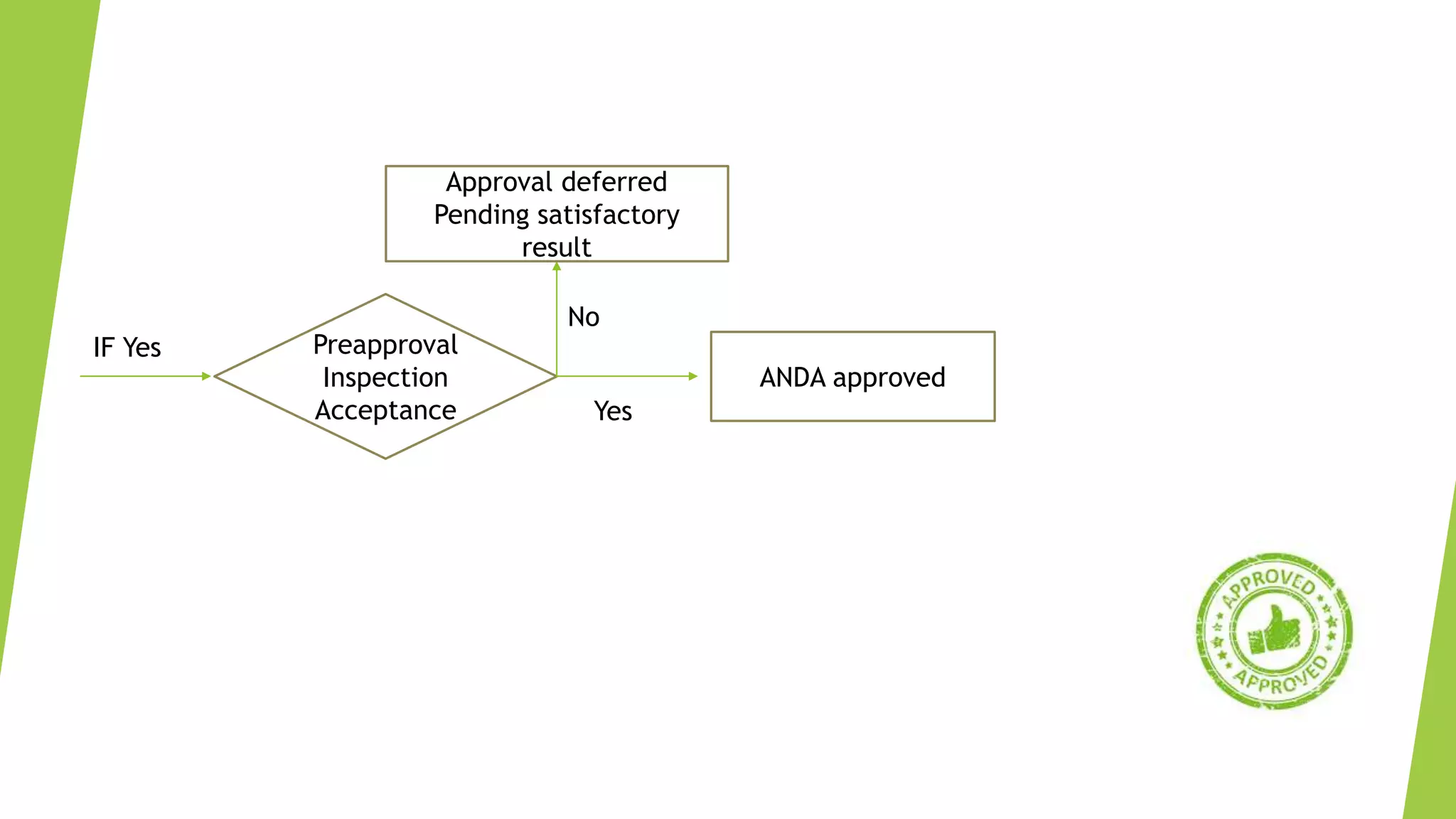








The document outlines the process and requirements for submitting an Abbreviated New Drug Application (ANDA) to the FDA for generic drug approval under the Hatch-Waxman Act, emphasizing the necessity to demonstrate bioequivalence to an existing licensed medication. It details the Common Technical Document (CTD) format, required application components, and distinctions between ANDA and New Drug Applications (NDA), including the data needed for evaluation. Additionally, the document addresses market exclusivity related to patents and various categories of exclusivity granted to innovator drugs.





























![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)

























































