Downloaded 455 times



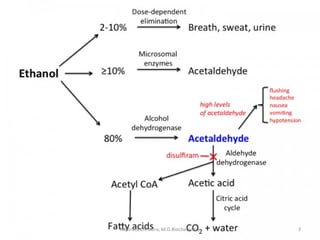

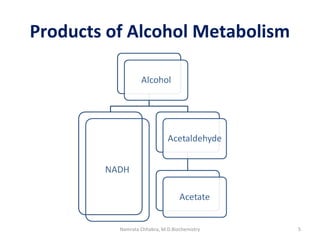





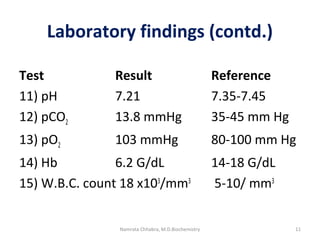




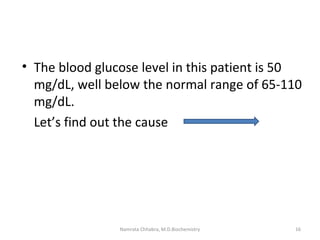


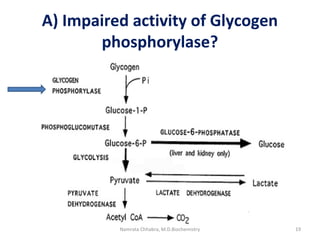
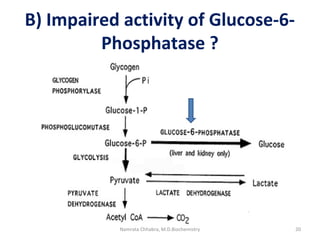
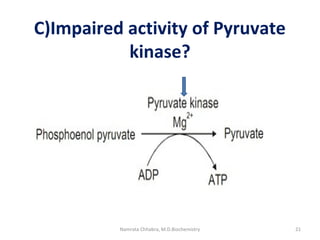
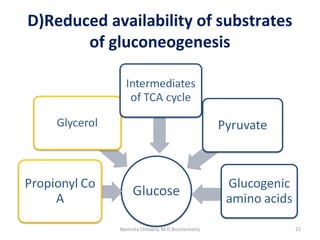

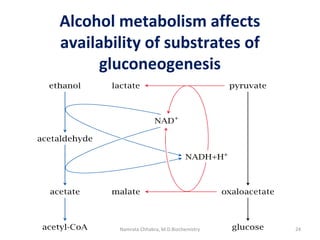
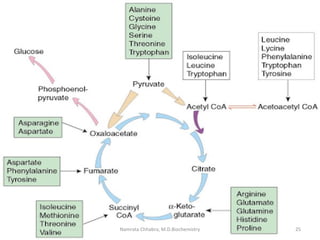
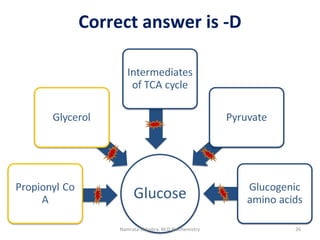

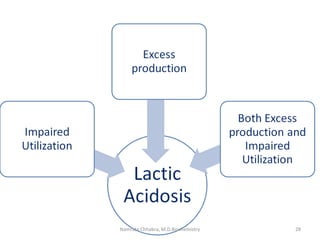

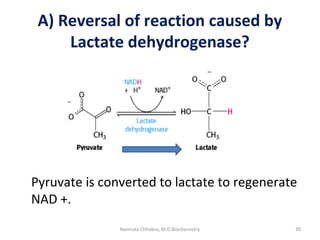
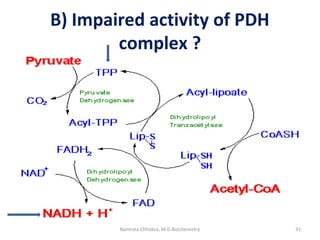
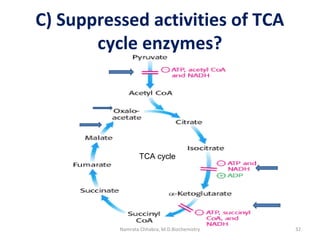

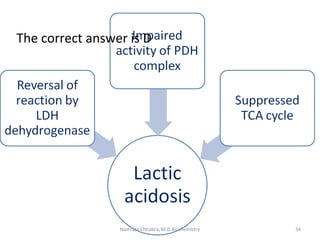







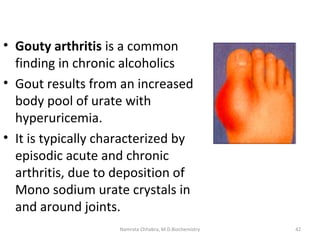



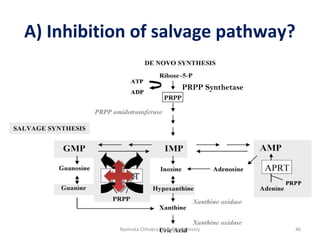
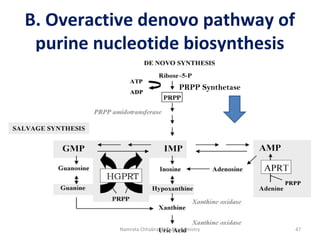
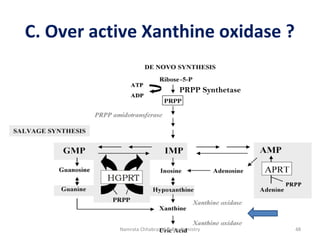
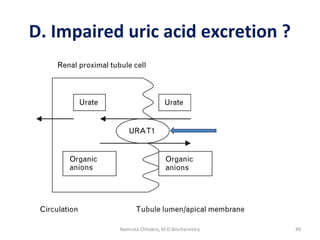

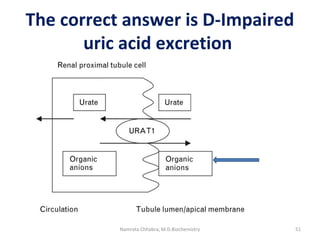

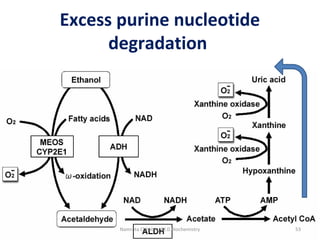







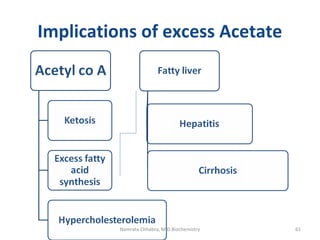

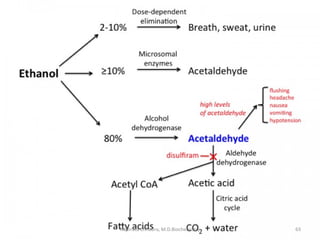


This document describes a case study of a 60-year-old male alcoholic who was brought to the hospital in serious condition. Laboratory tests revealed hypoglycemia, lactic acidosis, hyperuricemia, and other metabolic alterations. The patient's history of alcoholism and cirrhosis indicated that the metabolic derangements were due to alcohol-induced impairments in gluconeogenesis and tricarboxylic acid cycle function, leading to reduced glucose availability, increased lactate production, and impaired uric acid excretion. The patient was diagnosed with cirrhosis, portal hypertension, bleeding varices, sepsis, shock, renal failure, and other complications of long-term heavy alcohol use and malnutrition.


































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)








