

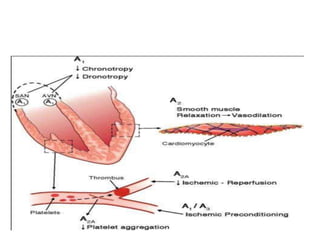


























![• Ischemic heart disease (IHD) is defined as a lack of oxygen and
decreased or no blood flow to the myocardium resulting from
coronary artery narrowing or obstruction.
• IHD may present as an acute coronary syndrome (ACS, which
includes unstable angina and non–ST-segment elevation or ST-
segment elevation myocardial infarction [MI]), chronic stable
exertional angina, ischemia without symptoms, or ischemia due to
coronary artery vasospasm (variant or Prinzmetal angina).
• The primary clinical manifestation of CHD is chest pain.](https://image.slidesharecdn.com/3anginapectoris-211001170802/85/3-angina-pectoris-30-320.jpg)
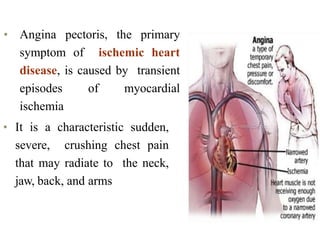







































![• Pharmacokinetic characteristics common to nitrates include a large
first pass effect of hepatic metabolism, short to very short half-lives
(except for isosorbide mononitrate [ISMN]), large volumes of
distribution, high clearance rates, and large interindividual variations
in plasma or blood concentrations.
• The half-life of nitroglycerin is 1 to 5 minutes regardless of the route,
hence the potential advantage of sustained-release and transdermal
products.
• Isosorbide dinitrate (ISDN) is metabolized to ISMN.
• ISMN has a half-life of about 5 hours and may be given once or twice
daily, depending on the product chosen.](https://image.slidesharecdn.com/3anginapectoris-211001170802/85/3-angina-pectoris-73-320.jpg)





















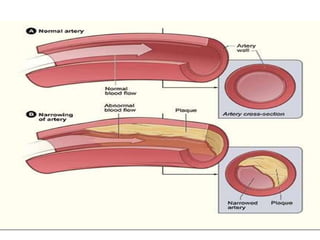
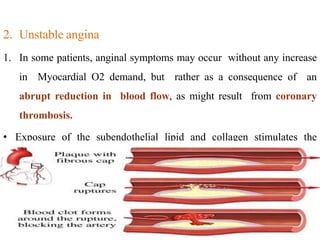
Coronary heart disease (CHD) is a condition characterized by impaired blood supply to the heart due to factors like atheroma and thrombosis, leading to myocardial ischemia and potential heart muscle damage. Epidemiological data indicate that CHD is a leading cause of death, particularly in developed countries, with risk factors including hypertension, smoking, diabetes, and psychological stress. The disease presents in various forms, including stable and unstable angina, and its underlying pathology involves progressive atherosclerosis and potential plaque rupture, necessitating clinical attention and management strategies.





































![CAD,_MI,_ANGINA,_CARDIOMYOPATHY[1] ppt.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/cadmianginacardiomyopathy1-240325064248-771a15bd-thumbnail.jpg?width=640&height=640&fit=bounds)

















































