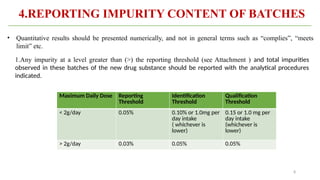
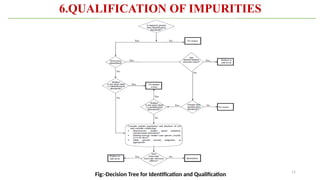
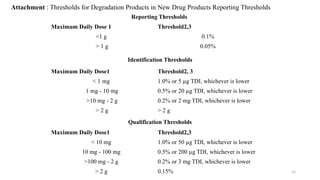
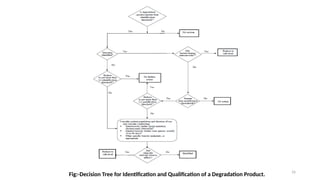
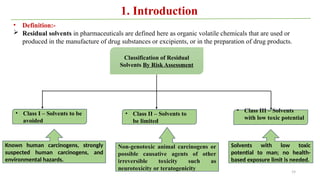
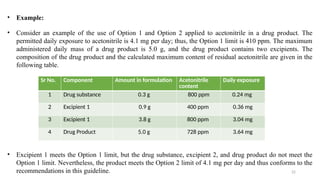
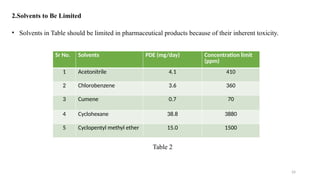
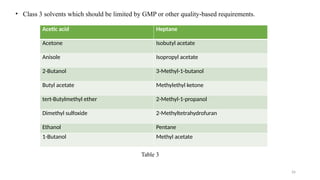
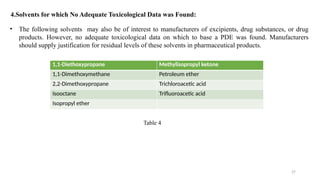
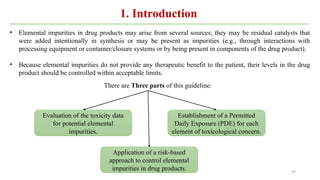
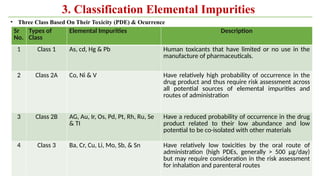
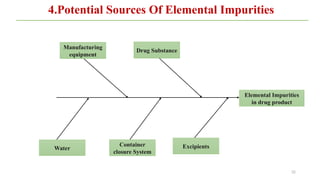
ICH Q3A guidelines focuses on the classification of impurities and establishes thresholds for control of impurities in API. ICH guidelines is for Impurities in New Drug Substances , New Drug Product. The ICH Q3C guideline addresses the control of residual solvents in drug substances and provides a list of solvents with their acceptable limits classified according to their toxicological properties. The ICH Q3 guidelines play a crucial role in ensuring the quality, safety, and efficacy of pharmaceutical and biotechnological products.