

![Von willebrand factor
• large multimeric glycoprotein -endothelial
cells and megakaryocytes
Storage –weibel-palade bodies[endothelium],
alpha granules[platelets]
Function-
1.carrier for factor VIII[degrades rapidly when
not bound ]
2.Acts as a ligand binds to platelet gpIb
• Cleavage -[ADAMTS13]-upshaw schulman
syndrome](https://image.slidesharecdn.com/bleedingandthrombosisfinal-180104183821/85/Bleeding-and-thrombosis-final-4-320.jpg)
![PLATELETS
• -Platelets from megakaryocytes ,
- normal – 1.5 to 4.5 lakhs
• life span of 7 to 10 days.
• Thrombopoeitin – liver .
-glycoproteins-
• GpIIb-IIIa –receptor of fibrinogen[glanzmanns
thrombasthenia]
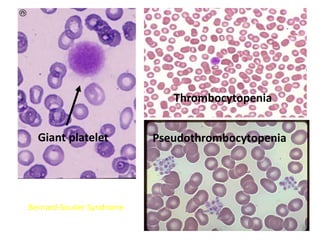
• GpIb-receptor of vWF[bernard-soulier
syndrome]](https://image.slidesharecdn.com/bleedingandthrombosisfinal-180104183821/85/Bleeding-and-thrombosis-final-5-320.jpg)
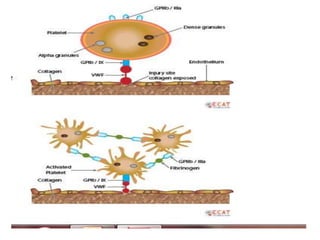
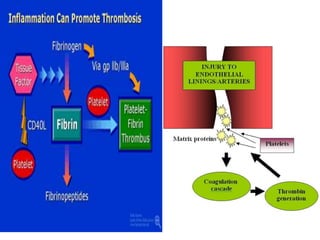
![COAGULATION FACTORS
• Coagulation factors are serine
proteases[enzymes],which acts by cleaving
proteins
• SYNTHESISED-
- Liver
- Endothelium
- Platelets](https://image.slidesharecdn.com/bleedingandthrombosisfinal-180104183821/85/Bleeding-and-thrombosis-final-7-320.jpg)
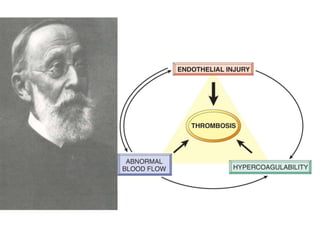
![Factors preventing coagulation.
Physiological – anticogulant mechanism-
• Anti-thrombin III-inhibits factor II,X
• Protein C-inactivates factor V,VIII
• Protein S-Cofactor for activated protein C
• Protein Z-degrades factor X
• Tissue Factor Pathway Inhibitor[TFPI]-neutralizes
factor X
• Thrombomodulin-thrombin,once bound to
thrombomodulin ,deactivates it.](https://image.slidesharecdn.com/bleedingandthrombosisfinal-180104183821/85/Bleeding-and-thrombosis-final-10-320.jpg)

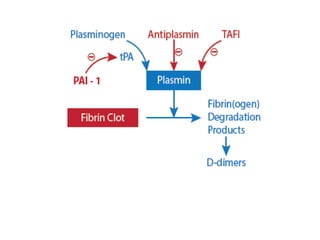
![FACTORS CONTROLLING FIBRINOLYSIS
• ALPHA2-ANTIPLASMIN-it inhibits plasmin
• Plasmin activator inhibitor-1&2-it inactivates
tissue plasminogen activator[tPA] and
urokinase](https://image.slidesharecdn.com/bleedingandthrombosisfinal-180104183821/85/Bleeding-and-thrombosis-final-12-320.jpg)



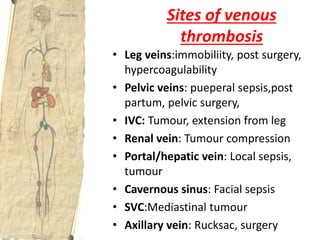


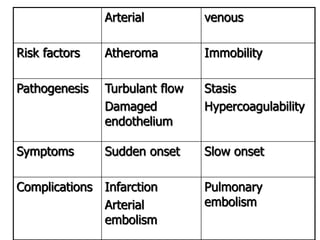


![Patient approach
• History
– major surgical procedures
– Trauma
– Recent hospitalization[within 90 days]
– Pregnancy
– smoking
• Drug history
– Oral contraceptives
– Hormone replacement therapy](https://image.slidesharecdn.com/bleedingandthrombosisfinal-180104183821/85/Bleeding-and-thrombosis-final-26-320.jpg)



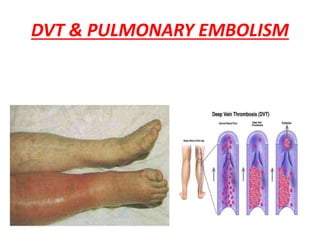

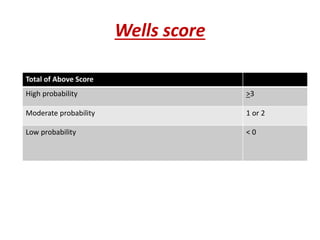
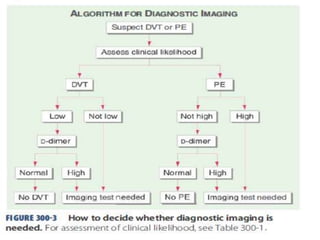

![• Rivaroxoban[factor X inhibitor]:approved as
monotherapy ,without parenteral bridging
anti-coagulant,no need for INR monitoring
start dose:15mg b.d x 3wks followed by
20 mg o.d
• Fondaparinux[anti Xa]:once daily,s.c inj
• Direct thrombin inhibitors:argatroban and
bivalirudin](https://image.slidesharecdn.com/bleedingandthrombosisfinal-180104183821/85/Bleeding-and-thrombosis-final-36-320.jpg)





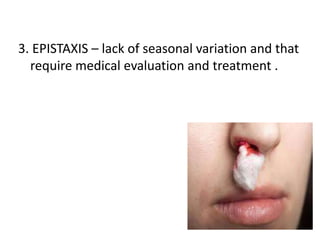


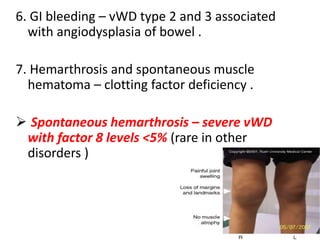














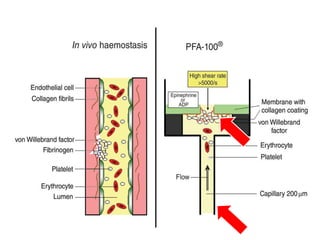
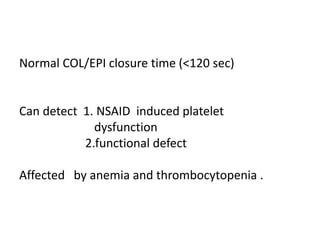





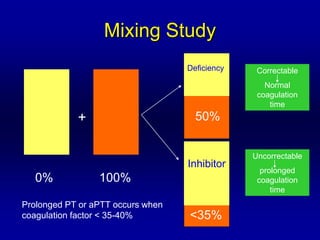






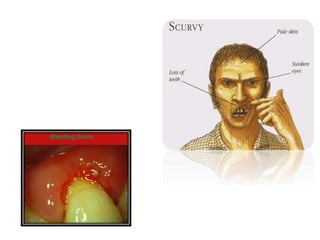









































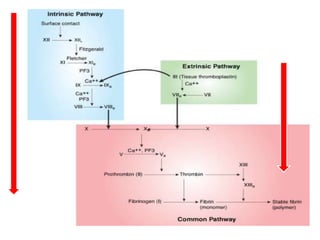
This document discusses the approach to disorders of bleeding and thrombosis. It covers the main components of hemostasis including the vascular endothelium, platelets, coagulation system, and fibrinolysis. It then discusses specific factors, disorders, investigations, and treatments related to bleeding and thrombotic disorders. The key points are that a thorough history, examination, and screening coagulation tests are needed to evaluate a patient. Additional targeted tests may be needed to identify the underlying cause, which could include a deficiency or inhibitor. Treatment depends on the specific disorder diagnosed.



































































