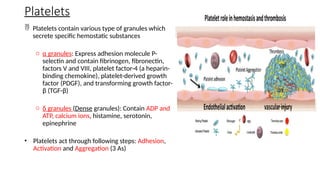
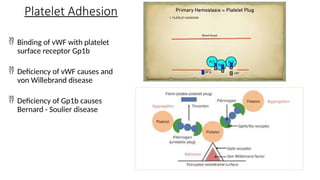
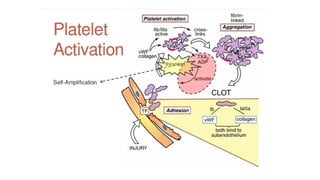
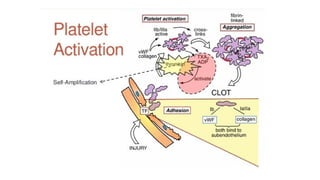
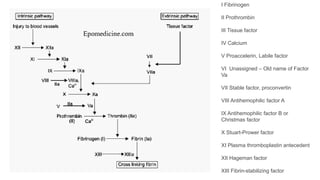
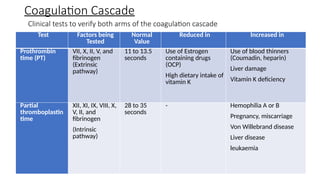
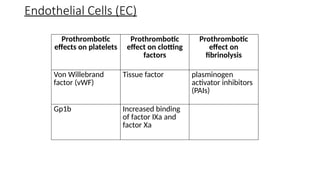
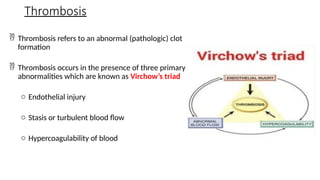
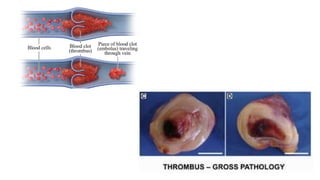
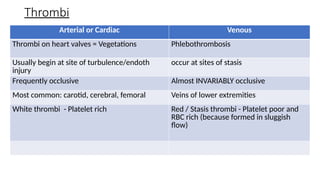
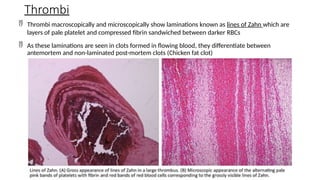
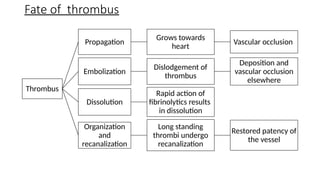
The document provides a comprehensive overview of hemostasis and thrombosis, defining key terms and processes involved in blood clot formation. It details the mechanisms of primary and secondary hemostasis, the roles of platelets and coagulation factors, and outlines Virchow's triad, which describes conditions leading to thrombosis. Clinical implications, including the significance of thrombi in lower limb deep vein thrombosis and potential risks, are also discussed.