Downloaded 13 times











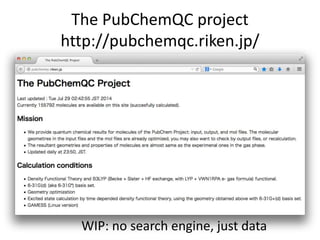
![Encoding molecule : SMILES
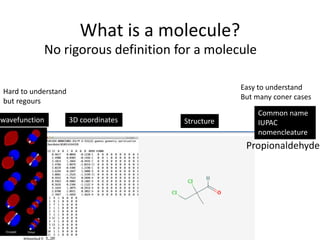
Encoding molecule
IUPAC nomenclature
tert-butyl N-[(2S,3S,5S)-5-[[4-[(1-benzyltetrazol-5-yl)
methoxy]phenyl]methyl]-3-hydroxy-6-[[(1S,2R)-
2-hydroxy-2,3-dihydro-1H-inden-1-yl]amino]-
6-oxo-1-phenylhexan-2-yl]carbamate
We can encode molecule
• SMILES
CN(C)CCOC12CCC(C3C1CCCC3)C4=CC=CC=C24
• InChI Made by IUPAC
InChI=1S/C20H29NO/c1-21(2)13-14-22-20-12-11
-15(16-7-3-5-9-18(16)20)17-8-4-6-10-19(17)20/
h3,5,7,9,15,17,19H,4,6,8,10-14H2,1-2H3
…
SMILES is a good encoding method for molecules](https://image.slidesharecdn.com/pubchemqcproject-141202185928-conversion-gate02/85/The-PubChemQC-Project-15-320.jpg)

![Example by SMILES
http://en.wikipedia.org/wiki/SMILES
分子構造SMILES
Nitrogen molecule N≡N N#N
copper sulfate Cu2+ SO42- [Cu+2].[O-]S(=O)(=O)[O-]
oenanthotoxin CCC[C@@H](O)CCC=CC=C
C#CC#CC=CCO
Vitamin B1 OCCc1c(C)[n+](=cs1)Cc2cnc(C
)nc(N)2
Aflatoxin B1 O1C=C[C@H]([C@H]1O2)c3c
2cc(OC)c4c3OC(=O)C5=C4CC
C(=O)5](https://image.slidesharecdn.com/pubchemqcproject-141202185928-conversion-gate02/85/The-PubChemQC-Project-17-320.jpg)
![Some corner cases
Two different SMILES for Ferrocene
• C12C3C4C5C1[Fe]23451234C5C1C2C3C45
• [CH-]1C=CC=C1.[CH-]1C=CC=C1.[Fe+2]](https://image.slidesharecdn.com/pubchemqcproject-141202185928-conversion-gate02/85/The-PubChemQC-Project-18-320.jpg)






[C@@H](O)[C@H](O)[C@@H](O)[C@
@H](O)1
– CCC[C@@H](O)CCC=CC=CC#CC#CC=CCO
– CC(=O)OCCC(/C)=CC[C@H](C(C)=C)CCC=C](https://image.slidesharecdn.com/pubchemqcproject-141202185928-conversion-gate02/85/The-PubChemQC-Project-27-320.jpg)
c3c2cc(OC)c4c3OC(=O)C5=C4CCC(=O)5
Ab initio calculation by
OpenBABEL](https://image.slidesharecdn.com/pubchemqcproject-141202185928-conversion-gate02/85/The-PubChemQC-Project-28-320.jpg)




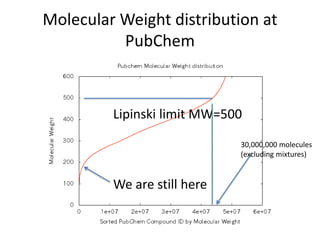






The PubChemQC project aims to create a comprehensive database of molecular properties through first-principles calculations, addressing the challenges of big data in chemistry. The project utilizes advanced computational methods, particularly density functional theory (DFT), to generate molecular data for potential drug candidates and aims for a Google-like search engine for chemistry. Despite the project's progress, including the processing of over a million molecules, the completion and comprehensive understanding of molecular data remain a significant challenge.