Download to read offline

















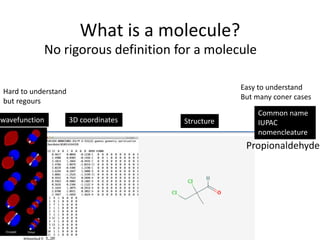

![Encoding molecule : SMILES
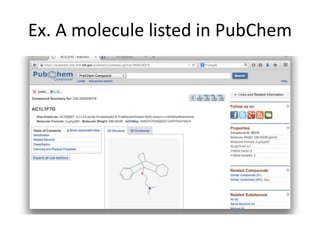
Encoding molecule
SMILES is a good encoding method for molecules
IUPAC nomenclature
tert-butyl N-[(2S,3S,5S)-5-[[4-[(1-benzyltetrazol-5-yl)
methoxy]phenyl]methyl]-3-hydroxy-6-[[(1S,2R)-
2-hydroxy-2,3-dihydro-1H-inden-1-yl]amino]-
6-oxo-1-phenylhexan-2-yl]carbamate
We can encode molecule
• SMILES
CN(C)CCOC12CCC(C3C1CCCC3)C4=CC=CC=C24
• InChI Made by IUPAC
InChI=1S/C20H29NO/c1-21(2)13-14-22-20-12-11
-15(16-7-3-5-9-18(16)20)17-8-4-6-10-19(17)20/
h3,5,7,9,15,17,19H,4,6,8,10-14H2,1-2H3
…](https://image.slidesharecdn.com/kobeworkshoppubchemqcproject-160217042216/85/Kobeworkshop-pubchemqc-project-22-320.jpg)

![Example by SMILES
http://en.wikipedia.org/wiki/SMILES
分子 構造 SMILES
Nitrogen molecule N≡N N#N
copper sulfate Cu2+ SO42- [Cu+2].[O-]S(=O)(=O)[O-]
oenanthotoxin CCC[C@@H](O)CCC=CC=C
C#CC#CC=CCO
Vitamin B1 OCCc1c(C)[n+](=cs1)Cc2cnc(C
)nc(N)2
Aflatoxin B1 O1C=C[C@H]([C@H]1O2)c3c
2cc(OC)c4c3OC(=O)C5=C4CC
C(=O)5](https://image.slidesharecdn.com/kobeworkshoppubchemqcproject-160217042216/85/Kobeworkshop-pubchemqc-project-24-320.jpg)
![Some corner cases
Two different SMILES for Ferrocene
• C12C3C4C5C1[Fe]23451234C5C1C2C3C45
• [CH-]1C=CC=C1.[CH-]1C=CC=C1.[Fe+2]](https://image.slidesharecdn.com/kobeworkshoppubchemqcproject-160217042216/85/Kobeworkshop-pubchemqc-project-25-320.jpg)






[C@@H](O)[C@H](O)[C@@H](O)[C@
@H](O)1
– CCC[C@@H](O)CCC=CC=CC#CC#CC=CCO
– CC(=O)OCCC(/C)=CC[C@H](C(C)=C)CCC=C](https://image.slidesharecdn.com/kobeworkshoppubchemqcproject-160217042216/85/Kobeworkshop-pubchemqc-project-34-320.jpg)
c3c2cc(OC)c4c3OC(=O)C5=C4CCC(=O)5
Ab initio calculation by
OpenBABEL](https://image.slidesharecdn.com/kobeworkshoppubchemqcproject-160217042216/85/Kobeworkshop-pubchemqc-project-35-320.jpg)





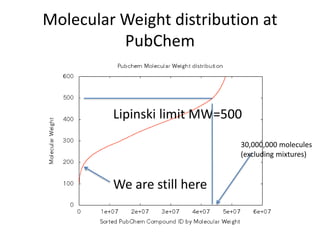





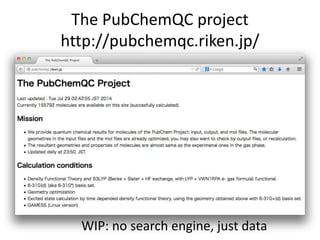
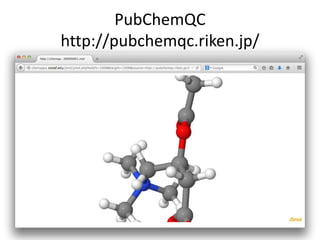
The PubChemQC project aims to create a comprehensive chemical database through first-principles calculations of molecular properties, enabling theoretical chemists to perform chemistry without physical experiments. This initiative addresses the complexity of electronic structure calculations and leverages vast computational resources and machine learning techniques to improve efficiency and accuracy in molecular property assessments. The project's objective is to construct a search engine-like platform for chemistry, facilitating better understanding and discovery within the vast chemical space.