材料は全て揃っている
• 基礎理論は大体ok
– “thefundamental laws necessary for the
mathematical treatment of a large part of physics
and the whole of chemistry are thus completely
known”
– Hartree Fock, DFT, MP2, … , PM3, PM6, AM1…
• 成熟した実装は多数あり、同じ結果を出す
– GAMESS, Gaussian, Qchem, Molpro….
• コンピューターリソース: 理研の施設
• データベース構築と、サービス提供
– 励起エネルギー、HOMO-LUMOギャップなどによる
「波動関数による」クエリー
• 化学的直感による補完、予測
aka. 機械学習
SMILESによる分子の符号化
Encoding molecule
SMILES isa good encoding method for molecules
IUPAC nomenclature
tert-butyl N-[(2S,3S,5S)-5-[[4-[(1-benzyltetrazol-5-yl)
methoxy]phenyl]methyl]-3-hydroxy-6-[[(1S,2R)-
2-hydroxy-2,3-dihydro-1H-inden-1-yl]amino]-
6-oxo-1-phenylhexan-2-yl]carbamate
We can encode molecule
• SMILES
CN(C)CCOC12CCC(C3C1CCCC3)C4=CC=CC=C24
…
11.
SMILESとは?
• Simplified MolecularInput Line Entry System
– ASCII文字を使った分子の符号化
– コンフォーマー、幾何異性体なども表現可能
– 人間にもプログラムからも読める
– だいたい「分子」と一対一対応する(universal SMILES
を用いることで)
• SMILESの発明者: David Weininger at USEPA Mid-Continent Ecology Division
Laboratory
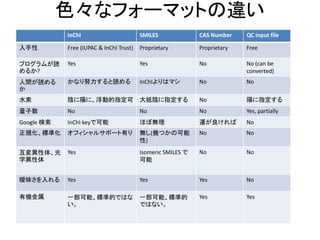
色々なフォーマットの違い
InChI SMILES CASNumber QC input file
入手性 Free (IUPAC & InChI Trust) Proprietary Proprietary Free
プログラムが読
めるか?
Yes Yes No No (can be
converted)
人間が読める
か
かなり努力すると読める InChIよりはマシ No No
水素 陰に陽に、浮動的指定可 大抵陰に指定する No 陽に指定する
量子数 No No No Yes, partially
Google 検索 InChI keyで可能 ほぼ無理 運が良ければ No
正規化、標準化 オフィシャルサポート有り 無し(幾つかの可能
性)
No No
互変異性体、光
学異性体
Yes Isomeric SMILES で
可能
No No
曖昧さを入れる Yes Yes Yes No
有機金属 一部可能、標準的ではな
い。
一部可能、標準的
ではない。
Yes Yes
Computer Resources
• RICC: Intel Xeon 5570 Westmere, 2.93GHz 8
cores/node) x 1000
– 1000-10000 molecules/day (MW 160)
– Heavily depend on conditions of other users
– Time limit: 8 hours
• Quest : Intel Core2 duo (1.6GHz/node) x 700
– 3000-8000 molecules / day (MW 160)
– 100-1000 molecules / day (MW 200-300)
– Time limit: 20 hours
• Some compounds fail to calculate are ignored for
this time.
38.
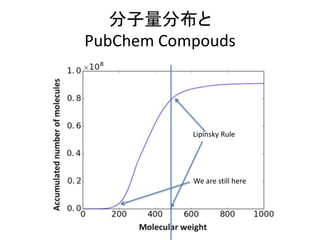
Molecular weight andLipinski Rule
• Lipinski’s five rule (Pfizer's rule of five): rule of
thumb for drug discovery
• No more than 5 hydrogen bond donors
• Not more than 10 hydrogen bond acceptors
• A molecular mass less than 500 daltons
• An octanol-water partition coefficient log P not greater than 5
• Molecular weight should be smaller than 500 is
very good for computational chemistry
– For routine calculations without experimental data
other than molecular formula
– If larger than 500, secondary or higher structure
becomes important. E.g., protein







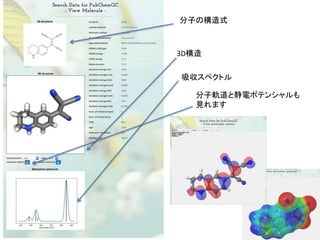
![SMILESによる分子の符号化
Encoding molecule
SMILES is a good encoding method for molecules
IUPAC nomenclature
tert-butyl N-[(2S,3S,5S)-5-[[4-[(1-benzyltetrazol-5-yl)
methoxy]phenyl]methyl]-3-hydroxy-6-[[(1S,2R)-
2-hydroxy-2,3-dihydro-1H-inden-1-yl]amino]-
6-oxo-1-phenylhexan-2-yl]carbamate
We can encode molecule
• SMILES
CN(C)CCOC12CCC(C3C1CCCC3)C4=CC=CC=C24
…](https://image.slidesharecdn.com/pubchemqc-170912042926/85/11-2017-9-17-Pubchemqc-10-320.jpg)

![Example by SMILES
http://en.wikipedia.org/wiki/SMILES
分子 構造 SMILES
Nitrogen molecule N≡N N#N
copper sulfate Cu2+ SO42- [Cu+2].[O-]S(=O)(=O)[O-]
oenanthotoxin CCC[C@@H](O)CCC=CC=C
C#CC#CC=CCO
Vitamin B1 OCCc1c(C)[n+](=cs1)Cc2cnc(C
)nc(N)2
Aflatoxin B1 O1C=C[C@H]([C@H]1O2)c3c
2cc(OC)c4c3OC(=O)C5=C4CC
C(=O)5](https://image.slidesharecdn.com/pubchemqc-170912042926/85/11-2017-9-17-Pubchemqc-12-320.jpg)
![SMILESで表現できない例
Ferroceneには二つの表現の仕方がある
• C12C3C4C5C1[Fe]23451234C5C1C2C3C45
• [CH-]1C=CC=C1.[CH-]1C=CC=C1.[Fe+2]](https://image.slidesharecdn.com/pubchemqc-170912042926/85/11-2017-9-17-Pubchemqc-13-320.jpg)



c3c2cc(OC)c4c3OC(=O)C5=C4CCC(=O)5
第一原理計算を
OpenBABEL
で](https://image.slidesharecdn.com/pubchemqc-170912042926/85/11-2017-9-17-Pubchemqc-21-320.jpg)
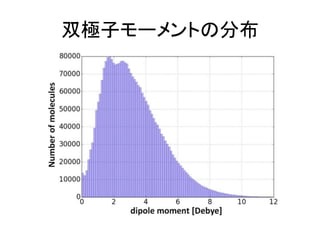
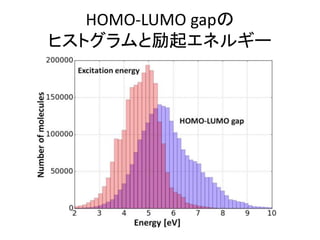
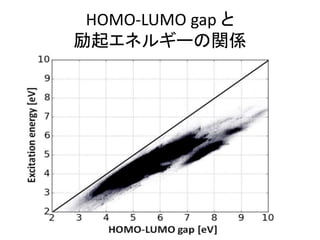



![機械学習と
HOMO-LUMO ギャップの予測
Method Kernel RMSE [eV]
SVM regression RBF 0.36
second-order polynomial 0.39
third-order polynomial 0.43
Ridge regression RBF 0.37
second-order polynomial 0.38
third-order polynomial 0.36
fourth-order polynomial 0.48](https://image.slidesharecdn.com/pubchemqc-170912042926/85/11-2017-9-17-Pubchemqc-30-320.jpg)

















![[DL輪読会]Deep Learning 第15章 表現学習](https://cdn.slidesharecdn.com/ss_thumbnails/deeplearning15-180601023904-thumbnail.jpg?width=640&height=640&fit=bounds)











![[DL輪読会]モデルベース強化学習とEnergy Based Model](https://cdn.slidesharecdn.com/ss_thumbnails/energybasedmodel-191129002008-thumbnail.jpg?width=640&height=640&fit=bounds)

































