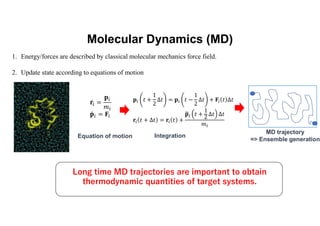
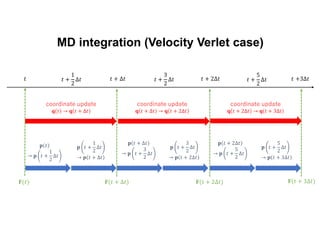
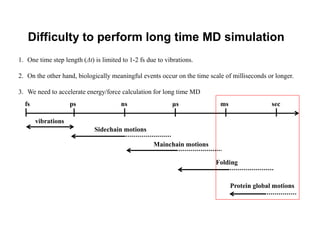
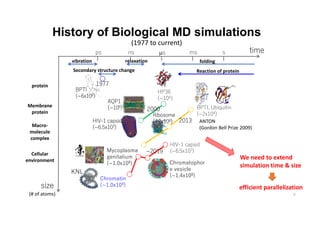
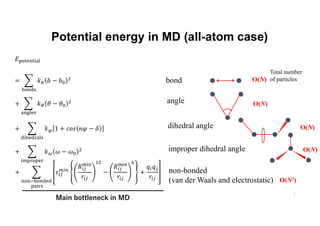
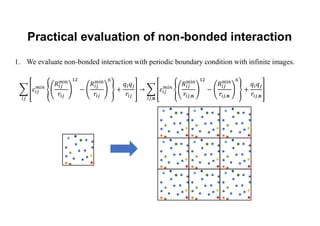
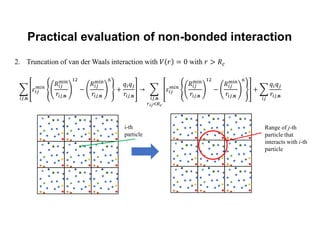
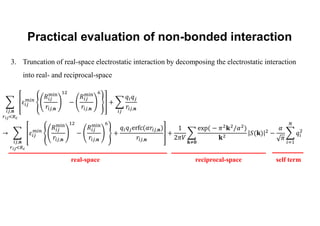
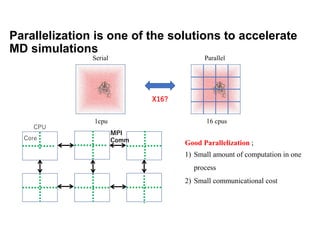
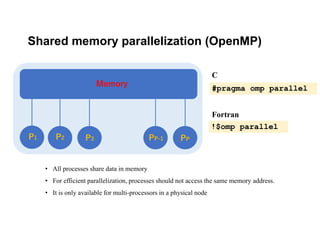
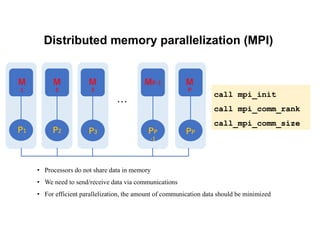
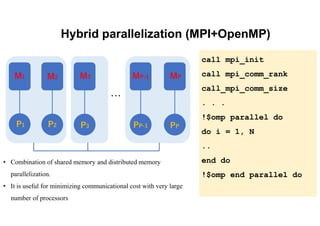
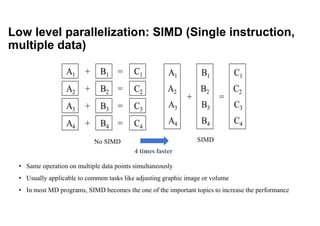
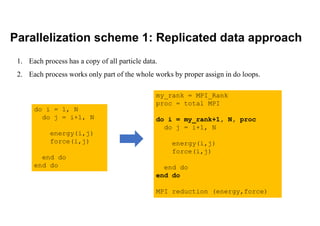
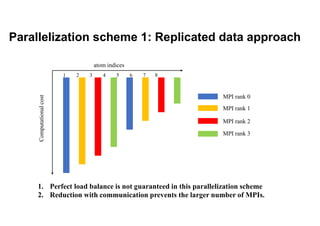
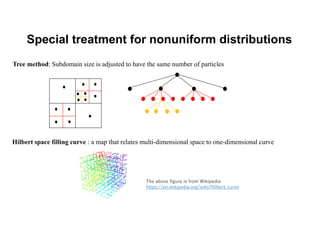
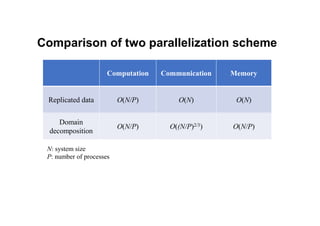
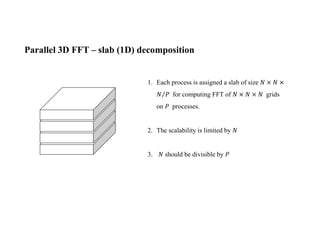
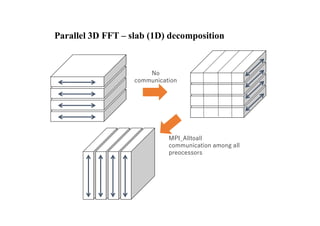
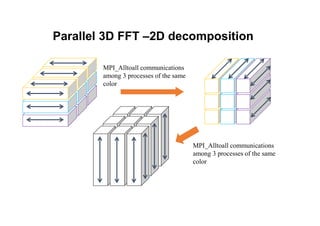
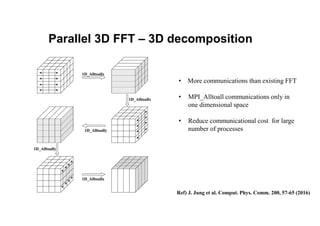
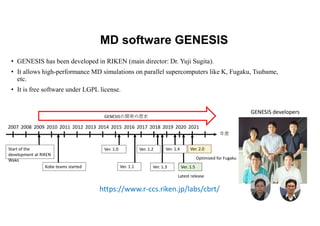
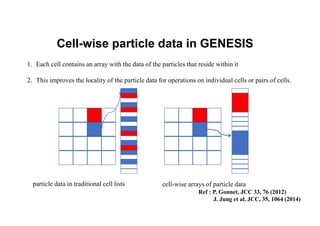
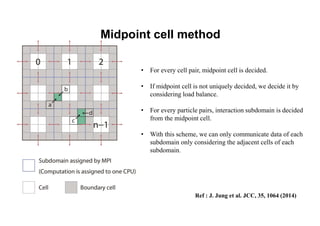
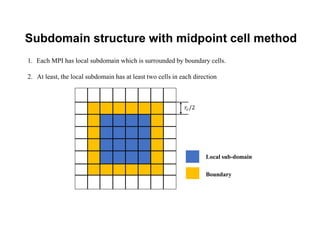
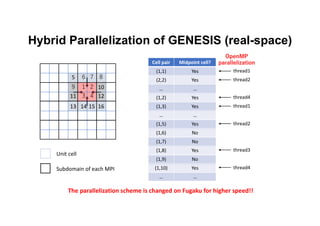
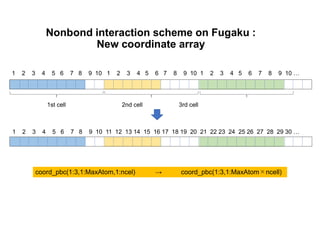
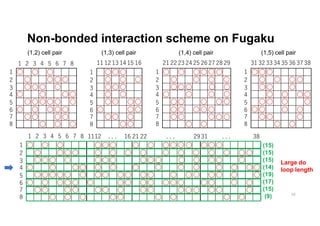
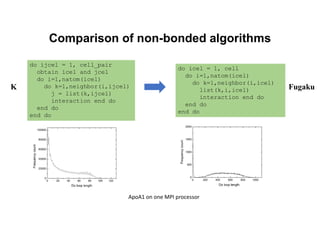
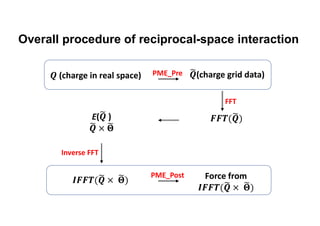
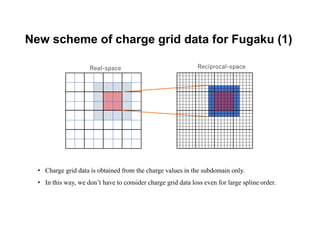
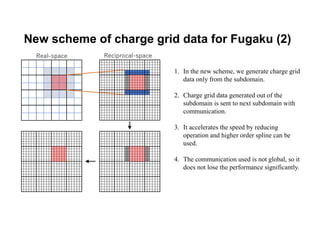
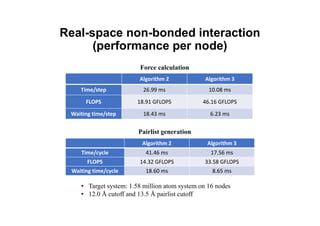
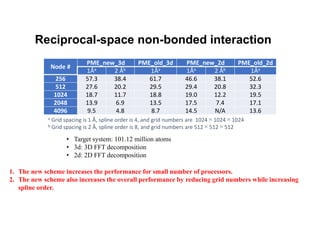
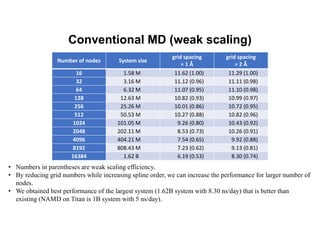
The document discusses parallelization techniques for molecular dynamics (MD) simulations, focusing on improving computation efficiency for long time MD simulations which face limitations due to short time steps and substantial biological events requiring longer time frames. It details various parallelization schemes such as replicated data approach, domain decomposition, and hybrid methods, as well as their associated advantages and challenges. Additionally, it references the implementation of these techniques in the Genesis MD software on the Fugaku supercomputer, emphasizing the optimization required to enhance performance.















































































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)
