Downloaded 107 times


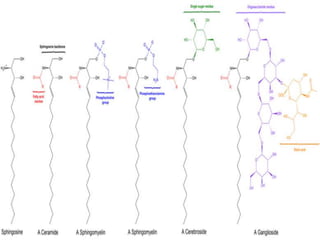

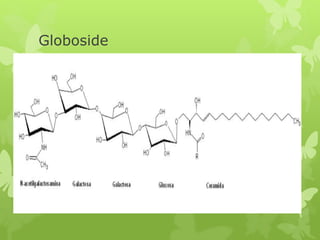
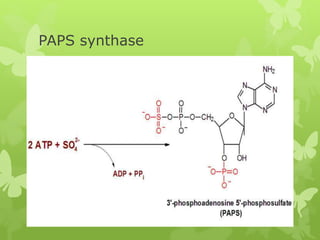


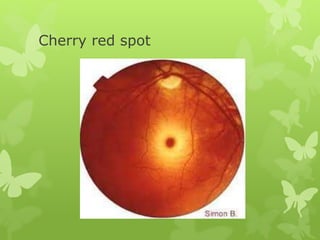

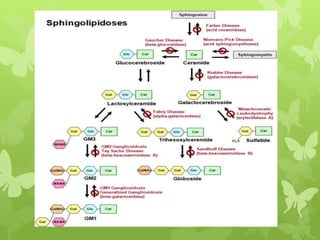
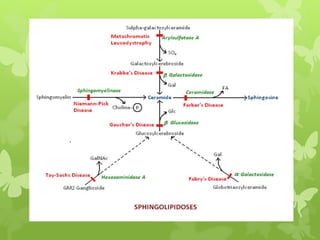

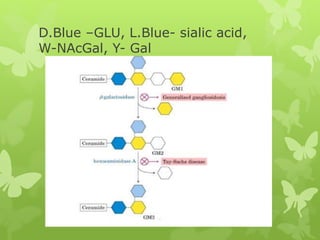

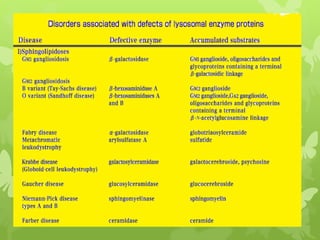
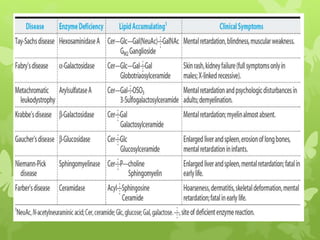
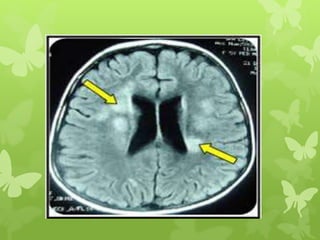


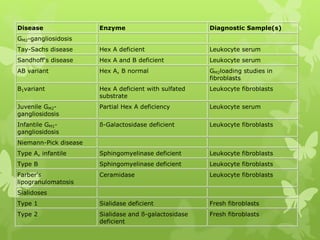


This document discusses sphingolipidoses, a class of rare genetic disorders caused by the accumulation of metabolites from complex lipids within neurons. Sphingolipidoses are characterized by progressive nervous system degeneration leading to symptoms like blindness, dementia, and paralysis. A cherry-red spot in the macula is a common sign seen during ophthalmologic examination, which can provide an important clue to diagnosis. Gangliosidoses are a type of sphingolipidosis caused by deficiencies in lysosomal enzymes involved in ganglioside metabolism, resulting in the storage of certain gangliosides in neurons and neurological deterioration. These disorders are ultimately fatal as there are currently no effective treatments available.











































































































