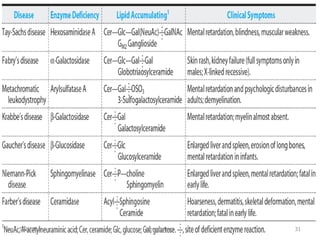
Lipid storage diseases are a group of inherited metabolic disorders caused by deficiencies of enzymes involved in metabolizing lipids. This leads to harmful accumulation of fatty materials called lipids in cells and tissues over time, damaging organs like the brain, liver, and bone marrow. The document discusses several specific lipid storage diseases including Gaucher disease, Niemann-Pick disease, Fabry disease, and others; their causes, signs and symptoms, methods of diagnosis, and available treatments are summarized for each one.